ARTÍCULO DE REVISIÓN
Andrés David-Ibarra¹, Juan Criado-Villamizar¹, Marco Niebles-Navas¹, Gabriel Rojas-Castro¹, Diana M. Monsalve¹, Yeny Acosta-Ampudia¹, Carolina Ramírez-Santana¹.
1. Centro de Estudio de Enfermedades Autoinmunes (CREA), Escuela de Medicina y Ciencias de la Salud, Universidad del Rosario, Bogotá, Colombia.
Recibido:
14 de marzo de 2024
Aceptado:
14 de marzo de 2024
Correspondencia:
DOI: 10.56050/01205498.2336
Resumen
Las enfermedades autoinmunes son trastornos crónicos caracterizados por la respuesta inmune del cuerpo contra sus propios tejidos, lo que causa inflamación y daño tisular. La inmunosenescencia, un proceso asociado al envejecimiento del sistema inmune, ha surgido como un factor crucial en el desarrollo y progresión de estas enfermedades. Los mecanismos de inmunosenescencia prematura compartidos por enfermedades autoinmunes reumatológicas, como la artritis reumatoide, el lupus eritematoso sistémico, las miopatías inflamatorias, la esclerosis sistémica, el síndrome de Sjögren y la vasculitis, están estrechamente relacionados con alteraciones específicas en la población de linfocitos T. Se observa una disminución significativa de linfocitos T vírgenes, que son fundamentales para la respuesta inmune adaptativa primaria, mientras que hay un aumento notable de linfocitos T de memoria efectora, que están implicados en respuestas inmunes secundarias y persistentes. Además, se produce una acumulación de células inmunosenescentes que presentan características de deterioro funcional y proliferativo. Estos cambios, junto con la disfunción mitocondrial, los cambios epigenéticos y el acortamiento de los telómeros, contribuyen de manera significativa a la instauración y mantenimiento de un fenotipo inflamatorio crónico, característico de las enfermedades autoinmunes. Comprender estos mecanismos es crucial para el desarrollo de nuevas estrategias terapéuticas que aborden tanto la autoinmunidad como la inmunosenescencia, especialmente dada la necesidad de tratamientos más efectivos en una población envejecida.
Palabras clave: Inmunosenescencia; Autoinmunidad; Inflamación; Enfermedades autoinmunes.
Abstract
Autoimmune diseases are chronic disorders characterized by the immune response against its own tissues, leading to inflammation and tissue damage. Immunosenescence, a process associated with aging of the immune system, has emerged as a crucial factor in the development and progression of these diseases. The mechanisms of premature immunosenescence shared by rheumatologic autoimmune diseases such as rheumatoid arthritis, systemic lupus erythematosus, inflammatory myopathies, systemic sclerosis, Sjögren’s syndrome, and vasculitis are closely related to specific alterations in the T lymphocyte population. There is a significant decrease in naïve T lymphocytes, which are essential for primary adaptive immune responses, while there is a notable increase in effector memory T lymphocytes, which are involved in secondary and persistent immune responses. Additionally, there is an accumulation of immunosenescent cells, which exhibit characteristics of functional and proliferative decline. These changes, along with mitochondrial dysfunction, epigenetic changes, and telomere shortening, significantly contribute to the establishment and maintenance of a chronic inflammatory phenotype, characteristic of autoimmune diseases. Understanding these mechanisms is crucial for the development of new therapeutic strategies that address both autoimmunity and immunosenescence, especially given the need for more effective treatments in an aging population.
Keywords: Immunosenescence; Autoimmunity; Inflammation; Autoimmune diseases.
Introducción
La inmunosenescencia es un término que describe el deterioro progresivo del sistema inmune relacionado con la edad, se caracteriza por remodelación de los tejidos inmunes y una disfunción en la respuesta inmune innata y adaptativa, manifestándose en una disminución de las respuestas proliferativas y la instauración de estados inflamatorios crónicos. Este fenómeno conlleva a una mayor susceptibilidad a enfermedades infecciosas, así como un aumento en la incidencia y severidad de las enfermedades autoinmunes (EAIs) (1,2).
Las EAIs son trastornos en los cuales el sistema inmune ataca erróneamente tejidos y órganos propios del cuerpo, provocando inflamación, daño tisular y disfunción orgánica. Estas enfermedades son una causa importante de morbilidad y mortalidad, afectando al 5-10 % de la población mundial (3). Múltiples factores como predisposición genética, factores ambientales y cambios en la regulación inmune similares a los observados durante la inmunosenescencia han sido asociados con el desarrollo de autoinmunidad (4,5). Las EAIs tienden a manifestarse con mayor frecuencia en ciertos grupos etarios. La edad representa un factor de riesgo significativo para la autoinmunidad, ya que muchas EAIs tienden a manifestarse con mayor frecuencia en la segunda mitad de la vida adulta. Durante este periodo, la respuesta inmune adaptativa a nuevos antígenos disminuye, al igual que la capacidad para mantener respuestas de linfocitos T (LT) de memoria (6,7). Estos cambios respaldan la hipótesis de la relación entre la inmunosenescencia y la autoinmunidad. Por esta razón, esta revisión busca explorar la interacción entre la inmunosenescencia y la autoinmunidad, así como conocer los mecanismos celulares y moleculares que contribuyen a la aparición y progresión de EAIs reumatológicas.
Mecanismos celulares y moleculares de inmunosenescencia
Los mecanismos asociados con la inmunosenescencia incluyen involución tímica, desregulación de células inmunes, disfunción mitocondrial, mecanismos epigenéticos, acortamiento de telómeros, inflammaging, entre otros (8–11).
El timo es el órgano responsable de la generación del repertorio de LT (12). El envejecimiento natural hace que el timo se atrofie progresivamente. La involución tímica relacionada con la edad se manifiesta a través de la alteración de la arquitectura del tejido y la reducción de la masa tímica, generando una disminución en el número de timocitos. Esto ocasiona menor producción de LT vírgenes que llegan a la periferia, una expansión clonal compensatoria de LT de memoria y reducción en la diversidad del repertorio de LT periféricos, comprometiendo la detección de patógenos (13).
A diferencia de las mutaciones somáticas esporádicas que ocurren en todo el genoma, el acortamiento de los telómeros es una característica constante del envejecimiento (14). Debido a la naturaleza altamente proliferativa de los LT tras su activación y la estimulación crónica a lo largo de la vida, se produce el acortamiento de telómeros (15). Aunque los LT pueden regular positivamente la telomerasa para mantener la longitud de sus telómeros, esta capacidad se ve afectada durante el envejecimiento, especialmente en los LT CD27-CD28-senescentes (16). Esto conduce a telómeros críticamente cortos con daño persistente del ADN (17).
La disfunción mitocondrial ocurre en la mayoría de los tejidos y células en humanos y ratones envejecidos (18). Aunque los LT de personas ancianas contienen más proteínas mitocondriales que los LT de individuos jóvenes, exhiben una fosforilación oxidativa alterada, sugiriendo que las mitocondrias disfuncionales se acumulan por un ineficiente reciclaje por autofagia (19). Las mitocondrias son centros de señalización que producen, transmiten y responden a especies reactivas de oxígeno (ROS) o picos de calcio (20). Estas vías de señalización están desreguladas en LT de ancianos (21). La disfunción mitocondrial en LT conduce a un fenotipo proinflamatorio, a través de mecanismos moleculares como la acumulación de metabolitos, alteraciones epigenéticas, modificaciones postranscripcionales y liberación de ADN mitocondrial (ADNmt) al citoplasma, que activa la vía cGAS-STING, generando la activación del inflamasoma y la transactivación de genes de citoquinas proinflamatorias (22). También se han descrito alteraciones del estado metabólico y la disfunción mitocondrial asociadas a la edad en monocitos y células dendríticas (DC) (23).
El epigenoma comprende diferentes niveles de alteración en la cromatina a través de la metilación del ADN y la modificación de histonas, asociados con cambios progresivos a la edad (24). Las alteraciones relacionadas con la edad en el epigenoma son diferentes entre los LT CD4+ y CD8+ (25), siendo más estable para los LT CD4+, mientras que los LT CD8+ vírgenes y de memoria muestran cambios en los patrones de accesibilidad a la cromatina hacia un estado más diferenciado y efector, que se ejemplifica por la represión de genes de la vía de señalización del receptor de interleuquina (IL)-7 (26). Otra característica epigenética del envejecimiento de los LT CD8+ es la inaccesibilidad a regiones regulatorias de genes involucrados en funciones celulares básicas, como los genes mitocondriales respiratorios (27). Este sesgo en los cambios epigenéticos puede explicar la pérdida preferencial de LT CD8+ vírgenes y la acumulación de LT CD8+ de memoria efectora CD45RA+ (TEMRA) con la edad. Se ha demostrado que la metilación del ADN es predictiva de la edad cronológica y fenotípica (28,29). La metilación del ADN en los LT coevoluciona con la activación del receptor de linfocitos T (TCR), la diferenciación y el compromiso de linaje (30). Adicionalmente, la heterogeneidad en la modificación de histonas en LT aumenta con la edad, lo que corresponde con una mayor variabilidad transcripcional (31).
Inmunidad Innata
La senescencia de las células madre hematopoyéticas (HSC) es la base de la inmunosenescencia, ya que se diferencian en varios tipos de células inmunes disfuncionales (32). La inflamación impide la actividad de autorenovación y acelera el envejecimiento de las HSC. La exposición a estímulos inflamatorios durante la edad temprana y media de la vida en ratones conduce al desarrollo de hemocitopenia en sangre periférica, citopenia de la médula ósea (MO) y acumulación de adipocitos en la MO, características típicas de hematopoyesis en ancianos (33). Los cambios de expresión se resumen en la Tabla 1 y 2.
Numerosos estudios han demostrado la importancia de las células B asociadas a la edad (ABC) como promotores de inmunosenescencia (35). Durante el envejecimiento, las ABC aumentan en número y disminuyen la respuesta inmune (36). Estas células se caracterizan por ser IgM+CD21-CD35-CD23- B220+CD19+ (37) y CD11c+B220+CD19+ (38). En LT, a medida que las personas envejecen, hay una disminución de células vírgenes y un aumento de memoria/efectora (39).
Inflammaging
La inflamación crónica de bajo grado, denominada “inflammaging”, es una característica distintiva del proceso de envejecimiento (45). El inflammaging se caracteriza por un aumento en los niveles de citoquinas proinflamatorias, como el factor de necrosis tumoral alfa (TNF-α), IL-6 e IL-1β, que contribuye a un medio proinflamatorio crónico. Este estado se asocia con el fenotipo secretor asociado a senescencia (SASP), donde las células senescentes secretan una variedad de factores inflamatorios y proteasas que promueven la inflamación crónica y el deterioro tisular (46). Se cree que varias causas contribuyen al desarrollo del inflammaging, incluyendo el estrés oxidativo, la acumulación de células senescentes, la disfunción mitocondrial y la activación crónica del sistema inmune debido a la exposición a antígenos a lo largo de la vida (47). La capacidad reducida de responder a nuevos antígenos, el acúmulo de células de memoria e inflammaging se consideran características distintivas de la inmunosenescencia.
A
continuación, se describirán los mecanismos celulares y moleculares
mediante los cuales la inmunosenescencia influye en la fisiopatología
y la presentación clínica de diferentes EAIs
(Figura 1).
|
Células |
Fenotipo |
Cambios |
|
Neutrófilos |
CD66b+/CD16+ |
|
|
Monocitos/macrófagos |
Clásicos (CD14++/CD16−) No clásicos (CD14+/ CD16++) Intermedios (CD14++/CD16+) |
|
|
Células dendríticas |
Mieloides (CD11c+ /CD123−) Plasmocitoides (CD11c− / CD123+) |
|
|
Natural Killer |
Citotóxicas (CD56low/CD16+) Secretoras de citoquinas (CD56high/CD16− ) |
|
Tabla 1. Cambios asociados a la edad en células del sistema inmune innato. Modificada de (34).
IFN: Interferón, IL: Interleuquina, mDCs: Células dendríticas mieloides, MHC: Complejo mayor de histocompatibilidad, NETs: Trampas extracelulares de neutrófilos, NK: Natural Killer, pDCs: Células dendríticas plasmocitoides, ROS: Especies reactivas de oxígeno, SASP: Fenotipo secretor asociado a senescencia, TLR: Receptor tipo Toll, TNF: Factor de necrosis tumoral.
|
Fenotipo |
Cambio |
Efecto fisiológico |
|
LB CD19+ |
Disminución |
|
|
LB vírgenes: CD19+/IgDHigh/IgMHigh /CD27− |
Disminución |
|
|
LB doble negativas CD19+/IgD−/(Switched Igs, IgG+/ IgA+/IgE+)/ CD27 |
Aumento |
|
|
LT CD4+/CD8+ vírgenes LT CD4+/CD45RA+/CCR7+ / CD27+/CD28+ LT CD8+/CD45RA+/CCR7+ / CD27+/CD28+ |
|
|
|
LT CD4+ TEMRA CD4+ /CD45RA+ / CCR7− /CD27− / CD28− LT CD8+ TEMRA CD8+ /CD45RA+ / CCR7− /CD27− / CD28− |
|
|
Tabla 2. Cambios asociados a la edad en células del sistema inmune adquirido. Modificada de (37,40–44).
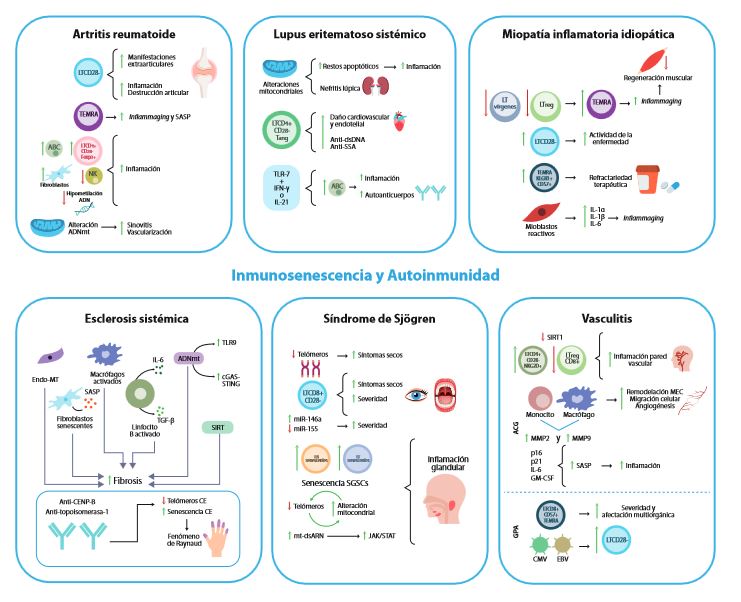
Figura 1. Interacción entre inmunosenescencia y enfermedades autoinmunes reumatológicas. Se ilustran los diferentes mecanismos de inmunosenescencia relacionados con la fisiopatología y la presentación clínica de diversas enfermedades autoinmunes reumatológicas, incluyendo artritis reumatoide, lupus eritematoso sistémico, miopatía inflamatoria idiopática, esclerosis sistémica, síndrome de Sjögren y vasculitis. Se destacan los roles de las células del sistema inmune innato y adaptativo en la liberación de citoquinas que contribuyen al fenotipo SASP e inflammaging, así como la influencia de la hipometilación del ADN, la presencia de microARNs, las alteraciones mitocondriales y el acortamiento de telómeros en la patogénesis de estas enfermedades.
ABC: Células B asociadas a la edad, ACG: Arteritis de células gigantes, ADNmt: ADN mitocondrial, Anti CENP-B: Anti-proteína centromérica B, Anti-dsDNA: Anti-DNA de doble cadena, Anti-SSA: Anti-antígeno A de Síndrome de Sjögren, CE: Célula Endotelial, CMV: Citomegalovirus, EBV: Virus de Epstein Barr, Endo-MT: Transición hacia fenotipo mesenquimatoso, GM-CSF: Factor estimulante de colonias de granulocitos, GPA: Granulomatosis con poliangeítis, IFN: Interferón, IL: Interleuquina, LTreg: Linfocito T regulador, MEC: Matriz extracelular, miR: Micro ARNs, MMP: Metaloproteinasas, mt-dsARN: ARN mitocondrial de doble cadena, NK: Natural Killer, SASP: Fenotipo secretor asociado a senescencia, SGSC: Células madre residentes en la glándula salival, SIRT: Sirtuina, Tang: Células T angiogénicas, TEMRA: Linfocitos T de memoria efectora CD45RA+, TGF: Factor de crecimiento transformante, TLR: Receptor tipo Toll.
Artritis Reumatoide
La Artritis reumatoide (AR) es una enfermedad inflamatoria, crónica y sistémica que afecta principalmente el tejido blando periarticular y las articulaciones, lo que conlleva a la destrucción del cartílago y el hueso (48). Aunque la AR puede desarrollarse a una edad temprana, la incidencia de esta enfermedad aumenta con la edad. La AR se asocia con varias características de envejecimiento acelerado, incluida la inmunosenescencia prematura y la presencia de comorbilidades (49).
La inmunosenescencia prematura en AR se establece por el deterioro de la capacidad regenerativa de LT. Los pacientes con AR exhiben una disminución en la frecuencia de los círculos de escisión del receptor de células T (TRECs), independientemente de la edad (50,51). Esta reducción ofrece una estimación indirecta de la integridad funcional del timo. Adicionalmente, en AR se ha observado una disminución en la producción tímica manifestada por una reducción de emigrantes tímicos recientes (RTEs) en circulación (LT CD31+ o TREC+). Esto podría ser debido a un suministro inadecuado de HSC desde la MO o a la involución tímica (52).
El repertorio de LT en pacientes con AR, se caracteriza por la expansión clonal de LT CD28- que son autorreactivos (51,53). La expansión de LT CD4+CD28- se observa tanto en pacientes con AR temprana como tardía principalmente en aquellos que portan el alelo de susceptibilidad del HLA-DRB1*04. Lo que sugiere que la inmunosenescencia acelerada puede ser impulsada genéticamente (54). Los LT CD28- contribuyen a la perpetuación de la inflamación y al empeoramiento de la AR al incrementar la producción de mediadores inflamatorios (55,56). En este contexto, Pieper y colaboradores demostraron diferencias funcionales entre los LT CD4+CD28- de sangre periférica y líquido sinovial. Los LT CD4+CD28- circulantes produjeron niveles más altos de interferón (IFN)-γ y TNF, mientras que los LT CD4+CD28- del sitio de la inflamación expresaron más CXCR3 y CCR6 e IL-17 (57). Adicionalmente, se observó que el estado de metilación del ADN y los niveles de ADN metiltransferasas en LT CD4+CD28- disminuyeron significativamente. Los genes sobreexpresados en respuesta a la hipometilación incluyeron aquellos con funciones efectoras como IFN-γ, CD70, KIR2DL4 y perforina, contribuyendo a la perpetuación de la inflamación (57,58).
Por otro lado, se ha observado que los pacientes con manifestaciones extraarticulares o con alteraciones ateroscleróticas mostraron una mayor prevalencia de LT senescentes, los cuales pueden ser de interés para posibles estrategias terapéuticas (56,59). Por ejemplo, el tratamiento con anti-TNF logró una reducción significativa de los LT CD8+CD28-, aunque no afectó los LT CD4+CD28-. Esta reducción se correlacionó con una mejora en la actividad de la enfermedad y los niveles de proteína C reactiva (60). Adicionalmente, los linfocitos TEMRA CD28- podrían contribuir al aumento de TNFα, IL-1β, IL-6 e IFN-γ (fenotipo SASP) favoreciendo el inflammaging y la severidad de la enfermedad (53,61). La AR también se asocia con la acumulación de ABCs (62,63), LT reguladores (LTregs) CD4+CD28-Foxp3+ (64) y fibroblastos sinoviales p16INK4a+ (65), que favorecen la inflamación y severidad de la enfermedad. Por otro lado, la actividad y número de natural killer (NK) están reducidas en pacientes con AR (66), impidiendo la eliminación adecuada de células senescentes implicadas en el daño tisular (67).
El acortamiento de telómeros está vinculado con mayor reactividad a autoantígenos y por tanto es un factor de riesgo para el desarrollo de AR. En pacientes con AR, se ha observado una erosión telomérica prematura en granulocitos y células mononucleares de sangre periférica (PBMCs) independiente de la edad (49,68). Esta erosión telomérica puede ser debido a una inflamación persistente, incremento del estrés oxidativo y expansión oligoclonal (49,54,69,70). También, se ha observado acortamiento de los telómeros de LT CD4+ de pacientes con AR por actividad defectuosa de la nucleasa reparadora de roturas del ADN MRE11A. Esta nucleasa se expresa en bajas concentraciones en pacientes con AR haciendo que los LT sean hipermotiles, invasivos y artritogénicos (71).
La disfunción mitocondrial en AR se caracteriza por una alta frecuencia de mutaciones en el ADN mitocondrial (ADNmt), favoreciendo la sinovitis, vascularización y altos niveles de TNF-α e IFN-γ en líquido sinovial (52,72). Se han encontrado altos niveles de ADNmt en plasma y líquido sinovial en AR, este último correlacionado con el factor reumatoide (73) y severidad de la enfermedad (46).
Es claro que la inmunosenescencia prematura puede contribuir al desarrollo o empeoramiento de la AR. Por tanto, estrategias terapéuticas novedosas dirigidas a controlar la presencia de células o mecanismos senescentes podrían ser de considerable valor en el tratamiento de esta enfermedad.
Lupus Eritematoso Sistémico
El lupus eritematoso sistémico (LES) es una enfermedad autoinmune sistémica y multifactorial (74). Los cambios metabólicos y funcionales provocan falla en la tolerancia periférica y aumento en la diferenciación de LT en fenotipos proinflamatorios exacerbando la enfermedad. El proceso continuo de activación inmune crónica y fenotipos proinflamatorios durante la historia natural del LES pueden conducir a la inmunosenescencia (75).
Las disfunciones mitocondriales en las células inmunes de LES, incluida la disminución de la capacidad redox, el aumento del estrés inflamatorio oxidativo, la apoptosis celular, el daño y la degradación del ADNmt facilita la inmunosenescencia (2,76). El LES se caracteriza por una activación anormal de LT, seguida de su muerte por suicidio, debido a la sobreproducción de ROS mitocondrial. Los LT exhiben una hiperpolarización mitocondrial y un agotamiento de ATP y glutatión que conducen a la muerte celular inducida por activación (77). Como consecuencia de la capacidad de eliminación defectuosa de las células apoptóticas, que se asocia con necrosis, los componentes celulares liberados estimulan los receptores tipo Toll (TLR) que provocan inflamación en LES (78). Además, el daño al ADNmt caracterizado por heteroplasmia D-310 y disminución del número de copias, está presente en LES (79). Las PBMCs y los neutrófilos en LES tienen mayor expresión de iniciadores de la apoptosis, caspasas 9 y 10, que se correlacionan positivamente con la expresión de la proteína de señalización antiviral mitocondrial y el factor regulador del interferón 7 (80). Se estableció que la disminución del número de copias de ADNmt y el aumento de los heteroplasmas D-310 se correlacionan con la nefritis lúpica (81). Además, la eliminación del ADN oxidativo D-310- 4977 y el ADNmt están correlacionados con los niveles séricos elevados de IL-10, IFN-α, IL-23, IFN-γ, IP-10 y MCP-1 en LES (82).
La longitud de los telómeros y la actividad de la telomerasa en PBMCs y granulocitos se han estudiado ampliamente en LES (83,84). La longitud de los telómeros de las PBMCs es menor en LES, especialmente en pacientes jóvenes. Sin embargo, la actividad de la telomerasa es mayor y se correlaciona con la actividad de la enfermedad, especialmente en linfocitos B (LB) (85).
Un análisis de biomarcadores inmunosenescentes en pacientes con LES reveló que el aumento de LT CD4+CD28- angiogénicos que contienen granzima B, perforina e IFN-γ, se correlacionan con daño cardiovascular y endotelial y con títulos de anticuerpos anti-DNA de doble cadena (dsDNA) y anti-SSA (86). Además, el aumento de la proporción IL-6/Factor de crecimiento transformante beta (TGF-β) impulsa la respuesta de los LT hacia la sobreexpresión de IL-22 e IL-17 en LES (87,88).
Las ABC se encuentran aumentadas en pacientes con LES (89). Estas células se diferencian a través de la coestimulación de TLR7 e IFN-γ o IL-21 (35). TLR7 es un factor crucial en LES, que regula los centros germinales (CG) y la producción de autoanticuerpos (90). El TLR7Y264H, aumenta el fenotipo asociado a la senescencia, incluida la acumulación de células ABC (B220+CD21-CX- CR5-CD19highCD11c+) y LB (CD19+CD95+B- CL6+) en LES (91).
En general, el acortamiento de telómeros, la disfunción mitocondrial, el cambio de proporción de LT y LB y el inflammaging son los posibles culpables de la inmunosenescencia prematura en LES.
Miopatías inflamatorias idiopáticas
Las miopatías inflamatorias idiopáticas (MII) son un grupo de condiciones autoinmunes caracterizadas por inflamación de músculos (miositis) y otros sistemas de órganos (piel, articulaciones, pulmones, corazón y tracto gastrointestinal), lo que resulta en disfunción orgánica generalizada y aumento de la morbilidad y mortalidad (92).
Los pacientes con miopatía por cuerpos de inclusión (MCI) presentan una disminución de LT vírgenes y LTregs, que son reemplazados gradualmente por LT de memoria efectora y linfocitos TEMRA, demostrando una proliferación e inmunovigilancia reducida y un aumento de las funciones efectoras. Estos cambios inmunosenescentes promueven el inflammaging y perjudican la regeneración muscular en miositis establecida (93,94).
Estudios histopatológicos han demostrado infiltrados de LT CD4+CD28- y LT CD8+CD28- en miopatías. Estas poblaciones expresan sustancias efectoras citotóxicas e inflamatorias (TNF e IFN-γ), correlacionado con la actividad de la enfermedad (95). Adicionalmente, estas células presentaron positividad para la proteína 1 de muerte celular programada (PD-1), conllevando a una acumulación de LT senescentes en tejido muscular afectado por miositis (96,97).
Adicionalmente, estos pacientes tienen una gran proporción de linfocitos CD8+CD28- TEMRA infiltrantes caracterizados por una regulación positiva de los receptores KLRG1 y CD57 de células NK (98,99). Se ha sugerido que el desarrollo de este fenotipo citotóxico de LT senescentes puede compensar las pérdidas observadas en LT y NK (100). Los linfocitos CD8+CD28- TEMRA, poseen una alta capacidad citotóxica y una capacidad proliferativa limitada, lo que las hace refractarias a las terapias inmunosupresoras tradicionales, siendo esta, una de las características clínicas relevantes en pacientes con miopatías (101).
Por último, se ha observado que pacientes con MIC o MCI presentan una acumulación de mutaciones en el ADNmt y una disfunción mitocondrial contribuyendo al deterioro de la función inmune (102). Adicionalmente, Morosetti y colaboradores encontraron que la tasa de proliferación y la clonogenicidad de mioblastos de MCI son significativamente más bajas y el tiempo de duplicación es más largo que los controles de la misma edad, observándose acortamiento en sus telómeros (103). Los mioblastos producen IL-1α, IL-1β, IL- 6, TNF-α y quimioquinas que ayudan al reclutamiento de leucocitos y a la interacción entre las células inflamatorias infiltrantes y las células musculares, favoreciendo la inflamación crónica (104).
El impacto negativo de la inmunosenescencia puede acelerar la atrofia muscular, debido a la insuficiencia del músculo para hacer frente al estrés celular continúo causado por la inflamación crónica. Por tanto, estudiar los mecanismos inmunosenescentes es de gran importancia para el conocimiento de la patogénesis de las miopatías y el posible desarrollo de intervenciones terapéuticas que ayuden al control de estas enfermedades.
Esclerosis sistémica
La esclerosis sistémica (ES) se caracteriza por disfunción inmune, inflamación crónica, daño endotelial vascular y fibrosis progresiva de la piel y órganos internos (105,106). Su origen es multifactorial, con variabilidad clínica, y un curso crónico y progresivo que resulta en discapacidad y alta mortalidad.
La acumulación de células senescentes e inmunosenescentes es importante en la patogénesis de la ES, por su fenotipo secretor que promueve la inflamación y la fibrosis. En biopsias de piel de pacientes con ES, se observó un aumento en el número de células senescentes (107,108). Sueros de pacientes con anticuerpos anti-centrómero (CENP)-B y anti-topoisomerasa-1 aceleran la senescencia de las células endoteliales (CE) vasculares, contribuyendo al fenómeno de Raynaud en ES. Estos autoanticuerpos reducen la producción de 6-ceto-prostaglandina-F1α y el contenido de telómeros, e incrementan la actividad de β-galactosidasa en CE. Estos hallazgos sugieren que estos autoanticuerpos pueden desempeñar un papel clave en la senescencia y el desarrollo de ES (109). Además, las CE en ES experimentan una transición hacia un fenotipo mesenquimatoso (Endo-MT), contribuyendo a la fibrosis y depósito de colágeno (110,111). Se ha observado una mayor cantidad de células Endo-MT en biopsias de piel de pacientes con ES (112,113). Los fibroblastos de estos pacientes muestran un patrón molecular relacionado con el procesamiento del ADN y el ARN, así como con procesos mitocondriales y metabólicos relacionados con mayor senescencia y menor autofagia y proliferación (108). Estos fibroblastos tienen mayores niveles de ROS intracelular, estrés oxidativo a nivel mitocondrial y producción de factores inflamatorios (114–116). El SASP en fibroblastos senescentes está relacionado con un ambiente fibrótico e inflamatorio (117,118).
La senescencia celular afecta a la inmunidad innata y adaptativa (119). Las DC tienen alteraciones en la presentación de antígenos y en la producción de IFN (120,121), y los neutrófilos presentan defectos funcionales (122). Además, se ha observado una disminución en la expresión de quimioquinas y receptores de activación de NK (123) y un fenotipo activado y profibrótico en macrófagos. La inmunosenescencia de LB en ES se caracteriza por un aumento de LB vírgenes y de memoria tardía, así como una disminución de LB de memoria activados (124). Además, los LB reguladores muestran un funcionamiento deteriorado con disminución de IL-10 (125). Estos cambios se acompañan de una mayor producción de IL-6 y TGF-β por parte de LB activados (126). En los LT CD4+, se observa una mayor activación de genes asociados a citoquinas y moléculas efectoras, y un aumento de LT CD27-CD28-, reflejando un estado de activación crónico (127). La relación CD4/CD8 aumenta en pacientes con ES (128), así como la expresión de CD57 (129). La expansión de LT CD8+CD28- en ES sugiere la presencia de LT senescentes con una replicación acelerada y telómeros más cortos.
La función tímica también se ve comprometida en ES. Los pacientes pediátricos tienen una reducción en RTEs y LT CD4+ vírgenes (130). Estudios han encontrado involución tímica incompleta asociada a la edad y a la probabilidad de desarrollar fibrosis pulmonar (131,132). El análisis del TCR revela menor variabilidad y presencia de LT oligoclonales expandidos, sugiriendo una respuesta inmune anómala (133,134). El trasplante autólogo de HSC mejora la diversidad del TCR y se relaciona con mejoría clínica (135), aunque esta respuesta varía entre individuos (136).
Los pacientes con ES muestran características prematuras de envejecimiento, incluyendo disfunción mitocondrial y acortamiento de telómeros. Se ha observado una disminución de mitocondrias funcionales en fibroblastos, junto con cambios morfológicos del ADNmt (137). Experimentos in vitro demostraron que el ADNmt estimula la activación de TLR9 y la vía cGAS-STING relacionados con la fisiopatología de la ES (138). Algunos estudios también sugieren un mayor acortamiento telomérico en pacientes con enfermedad pulmonar intersticial difusa y anticuerpos anti-topoisomerasa (114). Específicamente, se ha descrito acortamiento de telómeros en LT CD4+ vírgenes y de memoria central, así como en LT CD8+ de memoria efectora y LB de pacientes con ES (139). Además, algunos pacientes pueden presentar autoanticuerpos contra proteínas teloméricas asociados con enfermedad pulmonar grave (140,141).
En las últimas décadas, cada vez más estudios han explorado la contribución de las sirtuinas (SIRT) en la patogénesis de la ES. Las SIRT son desacetilasas de histonas con efectos en el metabolismo, la supervivencia celular y el envejecimiento (142,143). La reducción de la expresión de SIRT3 se observa en biopsias de piel de ES y fibroblastos, lo que sugiere un potencial terapéutico al aumentar SIRT3 para tratar la fibrosis asociada a ES. SIRT3 modula la señalización intracelular del TGF-β y mitiga las respuestas fibróticas en fibroblastos (144). Además, niveles séricos reducidos de SIRT1 y SIRT3 han sido detectados en pacientes con ES (145).
Todos los mecanismos de inmunosenescencia prematura descritos anteriormente, subrayan la complejidad de la ES y la necesidad de un enfoque multidisciplinario en su tratamiento y manejo clínico.
Síndrome de Sjögren
El síndrome de Sjögren (SS) es una enfermedad autoinmune crónica caracterizada por inflamación de las glándulas exocrinas, especialmente las salivales y lagrimales, causando sequedad en la boca y los ojos (146). Puede afectar también la piel, el sistema respiratorio y los genitales (147). La inmunosenescencia desempeña un papel crucial en el SS, contribuyendo a la patogénesis y la progresión de la enfermedad.
En modelos murinos de SS, la activación crónica del sistema inmune conduce a la acumulación de LT y LB senescentes en las glándulas salivales. Este fenómeno se debe al incremento en la expresión de la quimioquina CXCL13 por las células epiteliales, así como al aumento en la expresión del receptor CXCR5 en los linfocitos senescentes. Estos linfocitos producen un exceso de citoquinas proinflamatorias contribuyendo a la sialoadenitis, sugiriendo que podrían ser un objetivo terapéutico para tratar la xerostomía asociada con SS (148).
Por otro lado, se ha observado la pérdida de CD28 en LTCD8+ asociada a la gravedad de los síntomas secos y la actividad de la enfermedad sistémica (149). Los LT CD4+ vírgenes muestran una capacidad reducida de proliferación ex vivo e in vitro, con un fenotipo senescente caracterizado por telómeros acortados, reducción de IL-7R y acumulación de β-galactosidasa asociada a la senescencia (150).
La activación inicial de células epiteliales y células madre residentes en la glándula salival (SGSCs) por IFN-α, TNF y IL-6 conduce a la transición hacia un estado senescente (151–153). Este proceso, junto con la acumulación de linfocitos y mediadores inflamatorios, contribuye al daño celular (151). Además, la exposición a la radiación aumenta los marcadores de senescencia y SASP en las SGSCs (154). Pacientes con SS tienen un fenotipo senescente en SGSCs, caracterizado por una menor capacidad de regeneración y telómeros más cortos (153). Además, se ha visto una menor longitud telomérica en glándulas lagrimales (155).
La menor diversidad del TCR en SS se asocia con manifestaciones clínicas específicas y hallazgos de laboratorio como caries dental, trombocitopenia, colestasis hepática, anticuerpos antinucleares e hipergammaglobulinemia. Por el contrario, los pacientes con anticuerpos anti-SSA/SBB muestran una mayor diversidad del TCR (156).
En SS, se presenta un daño en las mitocondrias de las glándulas lagrimales, evidenciado por edema en las crestas mitocondriales y una menor longitud telomérica (155). Los cambios en las mitocondrias pueden aumentar la producción de ROS, lo que acorta los telómeros. A su vez, el acortamiento telomérico puede activar respuestas de daño del ADN, aumentando la producción de ROS y deteriorando la función mitocondrial (157). Estos hallazgos se correlacionan con una disminución en los niveles de ADNmt en células sanguíneas y una mayor cantidad de superóxido mitocondrial en pacientes con SS, lo que sugiere un vínculo entre el daño mitocondrial y la inflamación glandular (158). La presencia de ARN de doble cadena mitocondrial (mt-dsARN) se ha asociado con alteraciones exocrinas e inflamatorias en las glándulas salivales, y su exposición induce una mayor producción de mt- dsARN, activando la vía JAK/STAT y contribuyendo a la inflamación (159).
En PBMCs de pacientes con SS, se han encontrado niveles incrementados del microARN mir-146a y disminuidos del miR-155 (160). Estos microARN, conocidos como “inflamma-miR” en el envejecimiento, pueden ser marcadores útiles no solo de la inflamación, sino también de la inmunidad desregulada. La comprensión de todos estos mecanismos de inmunosenescencia puede proporcionar nuevas estrategias terapéuticas para el tratamiento del SS.
Vasculitis
La vasculitis es un grupo heterogéneo de enfermedades caracterizadas por inflamación y necrosis fibrinoide en las paredes de los vasos sanguíneos. La arteritis de células gigantes (ACG) es una enfermedad granulomatosa de grandes vasos en personas mayores de 50 años (161). Existe una superposición entre las vías implicadas en la inmunosenescencia y las observadas en la patogénesis de la ACG. La señalización de mTOR influye en la longevidad y el envejecimiento, esta vía es regulada por las SIRT (162). Se ha descrito que la señalización de mTORC1 juega un papel crucial en la inflamación y las lesiones vasculares en vasculitis, y su inhibición puede aumentar las LTregs y disminuir los LT efectores involucrados en la patogénesis de la enfermedad (163,164). En ACG, se demostró que el estrés oxidativo plasmático va acompañado de una expresión reducida de SIRT1 en PBMCs (165).
Los niveles de p16, p21, moléculas relacionadas con senescencia, IL-6 y factor estimulante de colonias de granulocitos (GM-CSF) están elevados en biopsias de arteria temporal en ACG (166). IL-6 y GM-CSF, parte del SASP, son blancos terapéuticos en ACG, lo que indica un papel relevante de la senescencia celular en la fisiopatología de la enfermedad (167,168).
En ACG, se observa una disminución de LT CD8+ vírgenes y un aumento de marcadores de senescencia en comparación con LT CD4+ (169). Se ha detectado un aumento de LT CD4+CD28- tanto en sangre periférica como en lesiones vasculares, con una regulación positiva del receptor NKG2D (170), que puede estimular la liberación de IFN-γ y GM-CSF, sugiriendo un papel patogénico en la inflamación vascular. La disminución del número y la función de LTregs inducida por el envejecimiento contribuye a la patogénesis de la enfermedad. En pacientes con ACG, los LTreg CD8+ muestran una reducción en su capacidad para suprimir LT efectores circundantes, debido a una vía de señalización aberrante a través de NOTCH4, lo que afecta la liberación de microvesículas que contienen NADPH oxidasa 2, importante en la supresión de la inflamación vascular (171).
Además, la inmunosenescencia se relaciona con la disfunción de las células mieloides, como macrófagos multinucleados clave en las lesiones granulomatosas de ACG (172). Estos pacientes muestran una alta expresión de enzimas asociadas a la inmunosenescencia vascular como metaloproteinasas (MMP)-2 y MMP-9 en monocitos y macrófagos (173). La activación de MMP-9 está vinculada con la remodelación de la matriz extracelular, la angiogénesis y la migración de células inmunes a la pared vascular, sugiriendo un papel crucial en la patogénesis de ACG.
En las arterias temporales de pacientes con ACG se han identificado cambios epigenéticos, como metilación del ADN relacionada con activación de la vía de calcineurina/NFAT (174) y sobreexpresión de miR-21, implicado en la diferenciación de LT de memoria y LT efectores (175). Estos cambios son similares a los observados en los LT senescentes (176).
En el caso de la poliangitis granulomatosa (GPA), una enfermedad autoinmune sistémica que se caracteriza por vasculitis necrotizante de vasos sanguíneos de tamaño pequeño a mediano y la presencia de anticuerpos anti-citoplasma de neutrófilos (177).
Varios estudios han confirmado una mayor frecuencia de LT CD28- en GPA (178–180). Además, se ha observado que LT de estos pacientes tienen telómeros más cortos (181), lo que indica una activación recurrente de LT. En pacientes menores de 40 años, un aumento en la frecuencia de LT CD8+TEMRA CD57+ circulantes se ha asociado con inflamación y severidad de la enfermedad (182). La inmunosenescencia en GPA también puede estar influenciada por la seropositividad al citomegalovirus y al virus de Epstein-Barr, lo que se asocia con una mayor frecuencia de LT CD28- y LT CD57+ (182). Además, las infecciones concomitantes con estos virus se han asociado con una pérdida de expresión de CD28 en LT CD8+ y LT CD4+ circulantes (179,180).
Para obtener una comprensión más completa del papel de la inmunosenescencia en vasculitis primarias es fundamental llevar a cabo análisis exhaustivos e integrados que consideren el impacto de las infecciones y los tratamientos.
Autoinmunidad y longevidad
La inmunosenescencia se asocia comúnmente con un aumento en la susceptibilidad o exacerbación de las EAIs. Sin embargo, los centenarios muestran una baja incidencia de EAIs. Este hecho plantea un enigma biológico interesante, ya que sugiere que la inmunosenescencia puede afectar de manera diferente a este grupo de individuos. Anaya y colaboradores (183) han propuesto varios mecanismos que podrían explicar este fenómeno: 1) La autoinmunidad natural parece ser más común en los centenarios que la autoinmunidad latente (184–186). 2) Los centenarios tienen una resiliencia inmune única, lo que significa que tienen la capacidad de mantener y restaurar rápidamente las funciones inmunes (187). 3) Los centenarios podrían tener una respuesta inflamatoria adaptada y remodelada que previene el envejecimiento sistémico y reduce la incidencia de EAIs (188). 4) La inmunosenescencia en centenarios podría ser un fenómeno adaptativo favorable, con una firma celular específica que minimiza la pérdida de actividad inmunológica. 5) La menor exposición a factores ambientales que hiperestimulan el sistema inmune podría proteger a los centenarios de desarrollar EAIs. 6) Los centenarios podrían tener un proceso de regulación de proteínas más eficiente, evitando la acumulación de proteínas defectuosas que contribuyen al envejecimiento (189). 7) Factores genéticos y epigenéticos podrían regular el metabolismo, la inflamación y la respuesta al estrés
en centenarios, lo que podría influir en su menor incidencia de EAIs (190). Aunque estas hipótesis ofrecen posibles explicaciones, se requiere más investigación para comprender completamente los mecanismos biológicos subyacentes a esta aparente resistencia a las EAIs en los centenarios.
Conclusiones
La inmunosenescencia es crucial en el desarrollo o exacerbación de las EAIs, caracterizándose por involución tímica, disfunción telomérica, estimulación antigénica crónica, disminución de LTregs, aumento de LT efectores, presencia de enfermedades infecciosas, producción de citoquinas proinflamatorias y alteraciones en moléculas solubles; contribuyendo a un ambiente inflamatorio crónico y al daño tisular asociado.
Esta influencia revela una brecha en la eficacia de los tratamientos actuales, no diseñados para abordar estas alteraciones. Su estudio podría conducir a nuevas estrategias terapéuticas para modular la función inmune en pacientes con EAIs, especialmente considerando el envejecimiento de la población.
Las terapias senolíticas, que eliminan células senescentes, emergen como una estrategia prometedora en EAIs, y han demostrado ser efectivas en modelos animales de AR y LES, reduciendo la inflamación y mejorando la función de los tejidos afectados.
Además, ciertas características asociadas a la inmunosenescencia, como la expresión de marcadores específicos o los cambios en la función celular, podrían utilizarse como biomarcadores en el diagnóstico y pronóstico de las EAIs. Explorar las correlaciones entre estos biomarcadores y las características clínicas específicas de cada enfermedad podría proporcionar información importante para la identificación de subgrupos de pacientes y el desarrollo de tratamientos personalizados.
Conflictos de interés
Ninguno.
Financiación
Este trabajado fue financiado por la Universidad del Rosario (ABN011).
Agradecimientos
Los autores agradecen a todos los miembros del CREA por sus contribuciones y fructíferos debates durante la preparación del manuscrito.
Referencias
Liu Z, Liang Q, Ren Y, Guo C, Ge X, Wang L, et al. Immunosenescence: molecular mechanisms and diseases. Signal Transduct Target Ther. 2023;8(1):200.
Montoya G. Immunosenescence. In: Autoimmunity: From Bench to Bedside Anaya JM, Shoenfeld Y, Villaraga A, Roger L, Cervera R. Bogotá: Universidad del Rosario; 2013. p. 185–201.
Ramos-Casals M, Brito-Zerón P, Kostov B, Sisó-Almirall A, Bosch X, Buss D, et al. Google-driven search for big data in autoimmune geoepidemiology: Analysis of 394,827 patients with systemic autoimmune diseases. Autoimmun Rev. 2015;14(8):670–9.
Zheng Y, Liu Q, Goronzy JJ, Weyand CM. Immune aging – A mechanism in autoimmune disease. Semin Immunol. 2023;69:101814.
Lindstrom TM, Robinson WH. Rheumatoid arthritis: a role for immunosenescence? J Am Geriatr Soc. 2010;58(8):1565–75.
Goronzy JJ, Weyand CM. Immune aging and autoimmunity. Cellular and Molecular Life Sciences. 2012;69(10):1615–23.
Liu Q, Zheng Y, Goronzy JJ, Weyand CM. T cell aging as a risk factor for autoimmunity. J Autoimmun. 2023;137:102947.
Nikolich-Žugich J. The twilight of immunity: emerging concepts in aging of the immune system. Nat Immunol. 2018;19(1):10–9.
Ucar D, Márquez EJ, Chung CH, Marches R, Rossi RJ, Uyar A, et al. The chromatin accessibility signature of human immune aging stems from CD8+ T cells. Journal of Experimental Medicine. 2017;214(10):3123–44.
Fulop T, Witkowski JM, Pawelec G, Alan C, Larbi A. On the Immunological Theory of Aging. In 2014. p. 163–76.
Rea IM, Gibson DS, McGilligan V, McNerlan SE, Alexander HD, Ross OA. Age and Age-Related Diseases: Role of Inflammation Triggers and Cytokines. Front Immunol. 2018;9.
Elyahu Y, Monsonego A. Thymus involution sets the clock of the aging T-cell landscape: Implications for declined immunity and tissue repair. Ageing Res Rev. 2021;65:101231.
Mittelbrunn M, Kroemer G. Hallmarks of T cell aging. Nat Immunol. 2021;22(6):687–98.
Blackburn EH, Greider CW, Szostak JW. Telomeres and telomerase: the path from maize, Tetrahymena and yeast to human cancer and aging. Nat Med. 2006;12(10):1133–8.
Sanderson SL, Simon AK. In aged primary T cells, mitochondrial stress contributes to telomere attrition measured by a novel imaging flow cytometry assay. Aging Cell. 2017;16(6):1234–43.
Plunkett FJ, Franzese O, Finney HM, Fletcher JM, Belaramani LL, Salmon M, et al. The Loss of Telomerase Activity in Highly Differentiated CD8+CD28− CD27− T Cells Is Associated with Decreased Akt (Ser473) Phosphorylation. The Journal of Immunology. 2007;178(12):7710–9.
Kell L, Simon AK, Alsaleh G, Cox LS. The central role of DNA damage in immunosenescence. Frontiers in Aging. 2023;4.
Desdín-Micó G, Soto-Heredero G, Aranda JF, Oller J, Carrasco E, Gabandé-Rodríguez E, et al. T cells with dysfunctional mitochondria induce multimorbidity and premature senescence. Science (1979). 2020;368(6497):1371–6.
Bektas A, Schurman SH, Gonzalez-Freire M, Dunn CA, Singh AK, Macian F, et al. Age-associated changes in human CD4+ T cells point to mitochondrial dysfunction consequent to impaired autophagy. Aging. 2019;11(21):9234–63.
Desdín-Micó G, Soto-Heredero G, Mittelbrunn M. Mitochondrial activity in T cells. Mitochondrion. 2018;41:51–7.
Quinn KM, Palchaudhuri R, Palmer CS, La Gruta NL. The clock is ticking: the impact of ageing on T cell metabolism. Clin Transl Immunology. 2019;8(11).
Soto-Heredero G, Gómez de las Heras MM, Gabandé-Rodríguez E, Oller J, Mittelbrunn M. Glycolysis – a key player in the inflammatory response. FEBS J. 2020;287(16):3350–69.
Chougnet CA, Thacker RI, Shehata HM, Hennies CM, Lehn MA, Lages CS, et al. Loss of Phagocytic and Antigen Cross-Presenting Capacity in Aging Dendritic Cells Is Associated with Mitochondrial Dysfunction. The Journal of Immunology. 2015;195(6):2624–32.
Sen P, Shah PP, Nativio R, Berger SL. Epigenetic Mechanisms of Longevity and Aging. Cell. 2016;166(4):822–39.
Hu B, Jadhav RR, Gustafson CE, Le Saux S, Ye Z, Li X, et al. Distinct Age-Related Epigenetic Signatures in CD4 and CD8 T Cells. Front Immunol. 2020;11.
Ucar D, Márquez EJ, Chung CH, Marches R, Rossi RJ, Uyar A, et al. The chromatin accessibility signature of human immune aging stems from CD8+ T cells. Journal of Experimental Medicine. 2017;214(10):3123–44.
Moskowitz DM, Zhang DW, Hu B, Le Saux S, Yanes RE, Ye Z, et al. Epigenomics of human CD8 T cell differentiation and aging. Sci Immunol. 2017;2(8).
Zhang Y, Wilson R, Heiss J, Breitling LP, Saum KU, Schöttker B, et al. DNA methylation signatures in peripheral blood strongly predict all-cause mortality. Nat Commun. 2017;8(1):14617.
Hannum G, Guinney J, Zhao L, Zhang L, Hughes G, Sadda S, et al. Genome-wide Methylation Profiles Reveal Quantitative Views of Human Aging Rates. Mol Cell. 2013;49(2):359–67.
Wilson CB, Makar KW, Shnyreva M, Fitzpatrick DR. DNA methylation and the expanding epigenetics of T cell lineage commitment. Semin Immunol. 2005;17(2):105–19.
Cheung P, Vallania F, Warsinske HC, Donato M, Schaffert S, Chang SE, et al. Single-Cell Chromatin Modification Profiling Reveals Increased Epigenetic Variations with Aging. Cell. 2018;173(6):1385-1397. e14.
Liggett LA, Sankaran VG. Unraveling Hematopoiesis through the Lens of Genomics. Cell. 2020;182(6):1384–400.
Bogeska R, Mikecin AM, Kaschutnig P, Fawaz M, Büchler-Schäff M, Le D, et al. Inflammatory exposure drives long-lived impairment of hematopoietic stem cell self-renewal activity and accelerated aging. Cell Stem Cell. 2022;29(8):1273-1284.e8.
Caruso C, Ligotti ME, Accardi G, Aiello A, Candore G. An immunologist’s guide to immunosenescence and its treatment. Expert Rev Clin Immunol. 2022;18(9):961–81.
Cancro MP. Age-Associated B Cells. Annu Rev Immunol. 2020;38(1):315–40.
Zhou D, Borsa M, Simon AK. Hallmarks and detection techniques of cellular senescence and cellular ageing in immune cells. Aging Cell. 2021;20(2).
Frasca D, Diaz A, Romero M, Garcia D, Blomberg BB. B Cell Immunosenescence. Annu Rev Cell Dev Biol. 2020;36(1):551–74.
Hao Y, O’Neill P, Naradikian MS, Scholz JL, Cancro MP. A B-cell subset uniquely responsive to innate stimuli accumulates in aged mice. Blood. 2011;118(5):1294–304.
Aiello A, Farzaneh F, Candore G, Caruso C, Davinelli S, Gambino CM, et al. Immunosenescence and Its Hallmarks: How to Oppose Aging Strategically? A Review of Potential Options for Therapeutic Intervention. Front Immunol. 2019;10:2247.
Xu W, Larbi A. Markers of T Cell Senescence in Humans. Int J Mol Sci. 2017;18(8):1742.
Teissier T, Boulanger E, Cox LS. Interconnections between Inflammageing and Immunosenescence during Ageing. Cells. 2022;11(3):359.
Frasca D, Diaz A, Romero M, Landin AM, Blomberg BB. Age effects on B cells and humoral immunity in humans. Ageing Res Rev. 2011;10(3):330–5.
Bulati M, Caruso C, Colonna-Romano G. From lymphopoiesis to plasma cells differentiation, the age-related modifications of B cell compartment are influenced by “inflamm-ageing.” Ageing Res Rev. 2017;36:125–36.
Brauning A, Rae M, Zhu G, Fulton E, Admasu TD, Stolzing A, et al. Aging of the Immune System: Focus on Natural Killer Cells Phenotype and Functions. Cells. 2022;11(6):1017.
Fulop T, Larbi A, Pawelec G, Khalil A, Cohen AA, Hirokawa K, et al. Immunology of Aging: the Birth of Inflammaging. Clin Rev Allergy Immunol. 2021;64(2):109–22.
Franceschi C, Garagnani P, Parini P, Giuliani C, Santoro A. Inflammaging: a new immune–metabolic viewpoint for age-related diseases. Nat Rev Endocrinol. 2018;14(10):576–90.
Fulop T, Larbi A, Dupuis G, Le Page A, Frost EH, Cohen AA, et al. Immunosenescence and Inflamm-Aging As Two Sides of the Same Coin: Friends or Foes? Front Immunol. 2018;8.
Di Matteo A, Bathon JM, Emery P. Rheumatoid arthritis. The Lancet. 2023;402(10416):2019–33.
Bauer ME. Accelerated immunosenescence in rheumatoid arthritis: impact on clinical progression. Immunity & Ageing. 2020;17(1):6.
Thewissen M, Somers V, Venken K, Linsen L, Van Paassen P, Geusens P, et al. Analyses of immunosenescent markers in patients with autoimmune disease. Clinical Immunology. 2007;123(2):209–18.
Thewissen M, Linsen L, Somers V. Premature Immunosenescence in Rheumatoid Arthritis and Multiple Sclerosis Patients. Ann N Y Acad Sci. 2005;1051(1):255–62.
Chalan P, van den Berg A, Kroesen BJ, Brouwer L, Boots A. Rheumatoid Arthritis, Immunosenescence and the Hallmarks of Aging. Curr Aging Sci. 2015;8(2):131–46.
Barbé-Tuana F, Funchal G, Schmitz CRR, Maurmann RM, Bauer ME. The interplay between immunosenescence and age-related diseases. Semin Immunopathol. 2020;42(5):545–57.
Schönland SO, Lopez C, Widmann T, Zimmer J, Bryl E, Goronzy JJ, et al. Premature telomeric loss in rheumatoid arthritis is genetically determined and involves both myeloid and lymphoid cell lineages. Proceedings of the National Academy of Sciences. 2003;100(23):13471–6.
Fasth AE, Snir O, Johansson AA, Nordmark B, Rahbar A, af Klint E, et al. Skewed distribution of proinflammatory CD4+CD28null T cells in rheumatoid arthritis. Arthritis Res Ther. 2007;9(5):R87.
Martens PB, Goronzy JJ, Schaid D, Weyand CM. Expansion of unusual CD4+ T cells in severe rheumatoid arthritis. Arthritis Rheum. 1997;40(6):1106–14.
Pieper J, Johansson S, Snir O, Linton L, Rieck M, Buckner JH, et al. Peripheral and Site-Specific <scp>CD</ scp> 4 + <scp>CD</scp> 28 null T Cells from Rheumatoid Arthritis Patients Show Distinct Characteristics. Scand J Immunol. 2014;79(2):149–55.
Liu Y, Chen Y, Richardson B. Decreased DNA methyltransferase levels contribute to abnormal gene expression in “senescent” CD4(+)CD28(-) T cells. Clin Immunol. 2009;132(2):257–65.
Gerli R, Schillaci G, Giordano A, Bocci EB, Bistoni O, Vaudo G, et al. CD4+CD28− T Lymphocytes Contribute to Early Atherosclerotic Damage in Rheumatoid Arthritis Patients. Circulation. 2004;109(22):2744–8.
Scarsi
M, Zigliogli T, Airó P. Decreased Circulating CD28-negative T Cells in
Patients with Rheumatoid Arthritis Treated with Abatacept Are
Correlated with Clinical Response. J Rheumatol. 2010;37(5):911–6.
Sallusto F, Lenig D, Förster R, Lipp M, Lanzavecchia A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature. 1999;401(6754):708–12.
Li Z yu, Cai ML, Qin Y, Chen Z. Age/autoimmunity-associated B cells in inflammatory arthritis: An emerging therapeutic target. Front Immunol. 2023;14.
Qin Y, Cai ML, Jin HZ, Huang W, Zhu C, Bozec A, et al. Age-associated B cells contribute to the pathogenesis of rheumatoid arthritis by inducing activation of fibroblast-like synoviocytes via TNF-α-mediated ERK1/2 and JAK-STAT1 pathways. Ann Rheum Dis. 2022;81(11):1504–14.
Fessler J, Raicht A, Husic R, Ficjan A, Schwarz C, Duftner C, et al. Novel Senescent Regulatory T-Cell Subset with Impaired Suppressive Function in Rheumatoid Arthritis. Front Immunol. 2017;8.
Del Rey MJ, Valín Á, Usategui A, Ergueta S, Martín E, Municio C, et al. Senescent synovial fibroblasts accumulate prematurely in rheumatoid arthritis tissues and display an enhanced inflammatory phenotype. Immunity & Ageing. 2019;16(1):29.
Shibatomi K, Ida H, Yamasaki S, Nakashima T, Origuchi T, Kawakami A, et al. A novel role for interleukin-18 in human natural killer cell death: high serum levels and low natural killer cell numbers in patients with systemic autoimmune diseases. Arthritis Rheum. 2001;44(4):884–92.
Sagiv A, Krizhanovsky V. Immunosurveillance of senescent cells: the bright side of the senescence program. Biogerontology. 2013;14(6):617–28.
Steer SE, Williams FMK, Kato B, Gardner JP, Norman PJ, Hall MA, et al. Reduced telomere length in rheumatoid arthritis is independent of disease activity and duration. Ann Rheum Dis. 2006;66(4):476–80.
Costenbader KH, Prescott J, Zee RY, De Vivo I. Immunosenescence and rheumatoid arthritis: Does telomere shortening predict impending disease? Autoimmun Rev. 2011;10(9):569–73.
Koetz K, Bryl E, Spickschen K, O’Fallon WM, Goronzy JJ, Weyand CM. T cell homeostasis in patients with rheumatoid arthritis. Proceedings of the National Academy of Sciences. 2000;97(16):9203–8.
Li Y, Shen Y, Hohensinner P, Ju J, Wen Z, Goodman SB, et al. Deficient Activity of the Nuclease MRE11A Induces T Cell Aging and Promotes Arthritogenic Effector Functions in Patients with Rheumatoid Arthritis. Immunity. 2016;45(4):903–16.
Da Sylva TR, Connor A, Mburu Y, Keystone E, Wu GE. Somatic mutations in the mitochondria of rheumatoid arthritis synoviocytes. Arthritis Res Ther. 2005;7(4):R844-51.
Hajizadeh S, DeGroot J, TeKoppele JM, Tarkowski A, Collins LV. Extracellular mitochondrial DNA and oxidatively damaged DNA in synovial fluid of patients with rheumatoid arthritis. Arthritis Res Ther. 2003;5(5):R234.
Tsokos GC. Systemic Lupus Erythematosus. New England Journal of Medicine. 2011;365(22):2110–21.
Suárez-Fueyo A, Bradley SJ, Tsokos GC. T cells in Systemic Lupus Erythematosus. Curr Opin Immunol. 2016;43:32–8.
Kurien BT, Scofield RH. Lipid peroxidation in systemic lupus erythematosus. Indian J Exp Biol. 2006;44(5):349–56.
Gergely P, Grossman C, Niland B, Puskas F, Neupane H, Allam F, et al. Mitochondrial hyperpolarization and ATP depletion in patients with systemic lupus erythematosus. Arthritis Rheum. 2002;46(1):175–90.
Kahlenberg JM, Kaplan MJ. The inflammasome and lupus: another innate immune mechanism contributing to disease pathogenesis? Curr Opin Rheumatol. 2014;26(5):475–81.
Li KJ, Wu CH, Hsieh SC, Lu MC, Tsai CY, Yu CL. Deranged bioenergetics and defective redox capacity in T lymphocytes and neutrophils are related to cellular dysfunction and increased oxidative stress in patients with active systemic lupus erythematosus. Clin Dev Immunol. 2012;2012:548516.
Su YJ, Cheng TT, Chen CJ, Chang WN, Tsai NW, Kung CT, et al. Investigation of the caspase-dependent mitochondrial apoptotic pathway in mononuclear cells of patients with systemic lupus erythematosus. J Transl Med. 2014;12(1):303.
Lee HT, Lin CS, Chen WS, Liao HT, Tsai CY, Wei YH. Leukocyte Mitochondrial DNA Alteration in Systemic Lupus Erythematosus and Its Relevance to the Susceptibility to Lupus Nephritis. Int J Mol Sci. 2012;13(7):8853–68.
Tsai CY. Oxidative DNA and mitochondrial DNA change in patients with SLE. Frontiers in Bioscience. 2017;22(3):4497.
Beier F, Balabanov S, Amberger CC, Hartmann U, Manger K, Dietz K, et al. Telomere length analysis in monocytes and lymphocytes from patients with systemic lupus erythematosus using multi-color flow- FISH. Lupus. 2007;16(12):955–62.
Wu CH, Hsieh SC, Li KJ, Lu MC, Yu CL. Premature telomere shortening in polymorphonuclear neutrophils from patients with systemic lupus erythematosus is related to the lupus disease activity. Lupus. 2007;16(4):265–72.
Kurosaka D, Yasuda J, Yoshida K, Yoneda A, Yasuda C, Kingetsu I, et al. Abnormal telomerase activity and telomere length in T and B cells from patients with systemic lupus erythematosus. J Rheumatol. 2006;33(6):1102–7.
López P, Rodríguez-Carrio J, Martínez-Zapico A, Caminal-Montero L, Suarez A. Senescent profile of angiogenic T cells from systemic lupus erythematosus patients. J Leukoc Biol. 2016;99(3):405–12.
Rekik
R, Smiti Khanfir M, Larbi T, Zamali I, Beldi-Ferchiou A, Kammoun O,
et al. Impaired TGF-β signaling in patients
with active systemic lupus erythematosus is associated with an
overexpression of IL-22. Cytokine. 2018;108:182–9.
van den Hoogen L, Sims G, van Roon J, Fritsch- Stork R. Aging and Systemic Lupus Erythematosus - Immunosenescence and Beyond. Curr Aging Sci. 2015;8(2):158–77.
Ruan P, Wang S, Yang M, Wu H. The ABC-associated immunosenescence and lifestyle interventions in autoimmune disease. Rheumatology and Immunology Research. 2022;3(3):128–35.
Fillatreau S, Manfroi B, Dörner T. Toll-like receptor signalling in B cells during systemic lupus erythematosus. Nat Rev Rheumatol. 2021;17(2):98–108.
Brown GJ, Cañete PF, Wang H, Medhavy A, Bones J, Roco JA, et al. TLR7 gain-of-function genetic variation causes human lupus. Nature. 2022;605(7909):349– 56.
Lundberg IE, Fujimoto M, Vencovsky J, Aggarwal R, Holmqvist M, Christopher-Stine L, et al. Idiopathic inflammatory myopathies. Nat Rev Dis Primers. 2021;7(1):86.
Strioga M, Pasukoniene V, Characiejus D. CD8+ CD28- and CD8+ CD57+ T cells and their role in health and disease. Immunology. 2011;134(1):17–32.
McLeish E, Slater N, Sooda A, Wilson A, Coudert JD, Lloyd TE, et al. Inclusion body myositis: The interplay between ageing, muscle degeneration and autoimmunity. Best Pract Res Clin Rheumatol. 2022;36(2):101761.
Fasth AER, Dastmalchi M, Rahbar A, Salomonsson S, Pandya JM, Lindroos E, et al. T Cell Infiltrates in the Muscles of Patients with Dermatomyositis and Polymyositis Are Dominated by CD28null T Cells. The Journal of Immunology. 2009;183(7):4792–9.
Knauss S, Preusse C, Allenbach Y, Leonard-Louis S, Touat M, Fischer N, et al. PD1 pathway in immune-mediated myopathies. Neurol Neuroimmunol Neuroinflamm. 2019;6(3).
Nelke C, Kleefeld F, Preusse C, Ruck T, Stenzel W. Inclusion body myositis and associated diseases: an argument for shared immune pathologies. Acta Neuropathol Commun. 2022;10(1):84.
Greenberg SA, Pinkus JL, Kong SW, Baecher-Allan C, Amato AA, Dorfman DM. Highly differentiated cytotoxic T cells in inclusion body myositis. Brain. 2019;142(9):2590–604.
Goyal NA, Coulis G, Duarte J, Farahat PK, Mannaa AH, Cauchii J, et al. Immunophenotyping of Inclusion Body Myositis Blood T and NK Cells. Neurology. 2022;98(13):e1374–83.
Vallejo AN, Mueller RG, Hamel DL, Way A, Dvergsten JA, Griffin P, et al. Expansions of NK-like αβT cells with chronologic aging: Novel lymphocyte effectors that compensate for functional deficits of conventional NK cells and T cells. Ageing Res Rev. 2011;10(3):354–61.
Greenberg SA. Inclusion body myositis: clinical features and pathogenesis. Nat Rev Rheumatol. 2019;15(5):257–72.
Lindgren U, Roos S, Hedberg Oldfors C, Moslemi AR, Lindberg C, Oldfors A. Mitochondrial pathology in inclusion body myositis. Neuromuscular Disorders. 2015;25(4):281–8.
Morosetti R, Broccolini A, Sancricca C, Gliubizzi C, Gidaro T, Tonali PA, et al. Increased aging in primary muscle cultures of sporadic inclusion-body myositis. Neurobiol Aging. 2010;31(7):1205–14.
De Rossi M, Bernasconi P, Baggi F, de Waal Malefyt R, Mantegazza R. Cytokines and chemokines are both expressed by human myoblasts: possible relevance for the immune pathogenesis of muscle inflammation. Int Immunol. 2000;12(9):1329–35.
Cutolo M, Soldano S, Smith V. Pathophysiology of systemic sclerosis: current understanding and new insights. Expert Rev Clin Immunol. 2019;15(7):753– 64.
Volkmann ER, Andréasson K, Smith V. Systemic sclerosis. The Lancet. 2023;401(10373):304–18.
Martyanov V, Whitfield ML, Varga J. Senescence Signature in Skin Biopsies From Systemic Sclerosis Patients Treated With Senolytic Therapy: Potential Predictor of Clinical Response? Arthritis & Rheumatology. 2019;71(10):1766–7.
Dumit
VI, Küttner V, Käppler J, Piera-Velazquez S, Jimenez SA,
Bruckner-Tuderman L, et al. Altered MCM Protein Levels and Autophagic
Flux in Aged and Systemic Sclerosis Dermal Fibroblasts. Journal of
Investigative Dermatology. 2014;134(9):2321– 30.
Shen CY, Li KJ, Lai PH, Yu CL, Hsieh SC. Anti-CENP-B and anti-TOPO-1-containing sera from systemic sclerosis-related diseases with Raynaud’s phenomenon induce vascular endothelial cell senescence not via classical p53-p21 pathway. Clin Rheumatol. 2018;37(3):749–56.
Romano E, Rosa I, Fioretto BS, Manetti M. The contribution of endothelial cells to tissue fibrosis. Curr Opin Rheumatol. 2024;36(1):52–60.
Di Benedetto P, Ruscitti P, Berardicurti O, Vomero M, Navarini L, Dolo V, et al. Endothelial-to-mesenchymal transition in systemic sclerosis. Clin Exp Immunol. 2021;205(1):12–27.
Manetti M, Romano E, Rosa I, Guiducci S, Bellando-Randone S, De Paulis A, et al. Endothelial-to-mesenchymal transition contributes to endothelial dysfunction and dermal fibrosis in systemic sclerosis. Ann Rheum Dis. 2017;76(5):924–34.
Chiu YH, Spierings J, van Laar JM, de Vries-Bouwstra JK, van Dijk M, Goldschmeding R. Association of endothelial to mesenchymal transition and cellular senescence with fibrosis in skin biopsies of systemic sclerosis patients: a cross-sectional study. Clin Exp Rheumatol. 2023;41(8):1612-1617
Usategui A, Municio C, Arias-Salgado EG, Martín M, Fernández-Varas B, Del Rey MJ, et al. Evidence of telomere attrition and a potential role for DNA damage in systemic sclerosis. Immun Ageing. 2022;19(1):7.
Tsou P, Talia NN, Pinney AJ, Kendzicky A, Piera-Velazquez S, Jimenez SA, et al. Effect of oxidative stress on protein tyrosine phosphatase 1B in scleroderma dermal fibroblasts. Arthritis Rheum. 2012;64(6):1978–89.
Sambo P, Baroni SS, Luchetti M, Paroncini P, Dusi S, Orlandini G, et al. Oxidative stress in scleroderma: Maintenance of scleroderma fibroblast phenotype by the constitutive up-regulation of reactive oxygen species generation through the NADPH oxidase complex pathway. Arthritis Rheum. 2001;44(11):2653–64.
Schafer MJ, White TA, Iijima K, Haak AJ, Ligresti G, Atkinson EJ, et al. Cellular senescence mediates fibrotic pulmonary disease. Nat Commun. 2017;8(1):14532.
Coppé JP, Desprez PY, Krtolica A, Campisi J. The Senescence-Associated Secretory Phenotype: The Dark Side of Tumor Suppression. Annual Review of Pathology: Mechanisms of Disease. 2010;5(1):99– 118.
Brown M, O’Reilly S. The immunopathogenesis of fibrosis in systemic sclerosis. Clin Exp Immunol. 2019;195(3):310–21.
Carvalheiro T, Zimmermann M, Radstake TRDJ, Marut W. Novel insights into dendritic cells in the pathogenesis of systemic sclerosis. Clin Exp Immunol. 2020;201(1):25–33.
Liu Q, Zaba LC, Satpathy AT, Longmire M, Zhang W, Li K, et al. Chromatin accessibility landscapes of skin cells in systemic sclerosis nominate dendritic cells in disease pathogenesis. Nat Commun. 2020;11(1):5843.
Impellizzieri D, Egholm C, Valaperti A, Distler O, Boyman O. Patients with systemic sclerosis show phenotypic and functional defects in neutrophils. Allergy. 2022;77(4):1274–84.
Benyamine A, Magalon J, Sabatier F, Lyonnet L, Robert S, Dumoulin C, et al. Natural Killer Cells Exhibit a Peculiar Phenotypic Profile in Systemic Sclerosis and Are Potent Inducers of Endothelial Microparticles Release. Front Immunol. 2018;9:1665.
Sato S, Fujimoto M, Hasegawa M, Takehara K. Altered blood B lymphocyte homeostasis in systemic sclerosis: Expanded naive B cells and diminished but activated memory B cells. Arthritis Rheum. 2004;50(6):1918–27.
Mavropoulos A, Simopoulou T, Varna A, Liaskos C, Katsiari CG, Bogdanos DP, et al. Breg Cells Are Numerically Decreased and Functionally Impaired in Patients With Systemic Sclerosis. Arthritis & Rheumatology. 2016;68(2):494–504.
Dumoitier N, Chaigne B, Régent A, Lofek S, Mhibik M, Dorfmüller P, et al. Scleroderma Peripheral B Lymphocytes Secrete Interleukin-6 and Transforming Growth Factor β and Activate Fibroblasts. Arthritis & Rheumatology. 2017;69(5):1078–89.
Paleja B, Low AHL, Kumar P, Saidin S, Lajam A, Nur Hazirah S, et al. Systemic Sclerosis Perturbs the Architecture of the Immunome. Front Immunol. 2020;11:1602.
Radstake TRDJ, van Bon L, Broen J, Hussiani A, Hesselstrand R, Wuttge DM, et al. The Pronounced Th17 Profile in Systemic Sclerosis (SSc) Together with Intracellular Expression of TGFβ and IFNγ Distinguishes SSc Phenotypes. PLoS One. 2009;4(6):e5903.
Maehara T, Kaneko N, Perugino CA, Mattoo H, Kers , Jesper, Allard-Chamard H, et al. Cytotoxic CD4+ T lymphocytes may induce endothelial cell apoptosis in systemic sclerosis. Journal of Clinical Investigation. 2020;130(5):2451–64.
Reiff A, Krogstad P, Moore S, Shaham B, Parkman R, Kitchen C, et al. Study of thymic size and function in children and adolescents with treatment refractory systemic sclerosis eligible for immunoablative therapy. Clinical Immunology. 2009;133(3):295–302.
Meunier M, Bazeli R, Feydy A, Drape JL, Kahan A, Allanore Y. Incomplete thymic involution in systemic sclerosis and rheumatoid arthritis. Joint Bone Spine. 2013;80(1):48–51.
Ferri C, Colaci M, Battolla L, Giuggioli D, Sebastiani M. Thymus alterations and systemic sclerosis. Rheumatology. 2006;45(1):72–5.
Servaas NH, Zaaraoui-Boutahar F, Wichers CGK, Ottria A, Chouri E, Affandi AJ, et al. Longitudinal analysis of T-cell receptor repertoires reveals persistence of antigen-driven CD4+ and CD8+ T-cell clusters in systemic sclerosis. J Autoimmun. 2021;117:102574.
Sakkas LI, Xu B, Artlett CM, Lu S, Jimenez SA, Platsoucas CD. Oligoclonal T Cell Expansion in the Skin of Patients with Systemic Sclerosis. The Journal of Immunology. 2002;168(7):3649–59.
Arruda LCM, Malmegrim KCR, Lima-Júnior JR, Clave E, Dias JBE, Moraes DA, et al. Immune rebound associates with a favorable clinical response to autologous HSCT in systemic sclerosis patients. Blood Adv. 2018;2(2):126–41.
Farge D, Arruda LCM, Brigant F, Clave E, Douay C, Marjanovic Z, et al. Long-term immune reconstitution and T cell repertoire analysis after autologous hematopoietic stem cell transplantation in systemic sclerosis patients. J Hematol Oncol. 2017;10(1):21.
Zhou X, Trinh-Minh T, Tran-Manh C, Gießl A, Bergmann C, Györfi A, et al. Impaired Mitochondrial Transcription Factor A Expression Promotes Mitochondrial Damage to Drive Fibroblast Activation and Fibrosis in Systemic Sclerosis. Arthritis & Rheumatology. 2022;74(5):871–81.
Ryu C, Walia A, Ortiz V, Perry C, Woo S, Reeves BC, et al. Bioactive Plasma Mitochondrial DNA Is Associated With Disease Progression in Scleroderma Associated Interstitial Lung Disease. Arthritis & Rheumatology. 2020;72(11):1905–15.
Park JA, Lee J, Kim HR, Fujii H, Weyand CM, Goronzy JJ, et al. AB0050 Immunosenescence of T and B cells in systemic sclerosis. Ann Rheum Dis. 2013;71(Suppl 3):640.10-640.
Adler BL, Boin F, Wolters PJ, Bingham CO, Shah AA, Greider C, et al. Autoantibodies targeting telomere-associated proteins in systemic sclerosis. Ann Rheum Dis. 2021;80(7):912–9.
Lakota K, Varga J. Linking autoimmunity, short telomeres and lung fibrosis in SSc. Nat Rev Rheumatol. 2021;17(9):511–2.
Lee SH, Lee JH, Lee HY, Min KJ. Sirtuin signaling in cellular senescence and aging. BMB Rep. 2019;52(1):24–34.
Grabowska W, Sikora E, Bielak-Zmijewska A. Sirtuins, a promising target in slowing down the ageing process. Biogerontology. 2017;18(4):447–76.
Akamata K, Wei J, Bhattacharyya M, Cheresh P, Bonner MY, Arbiser JL, et al. SIRT3 is attenuated in systemic sclerosis skin and lungs, and its pharmacologic activation mitigates organ fibrosis. Oncotarget. 2016;7(43):69321–36.
Manetti M, Rosa I, Fioretto BS, Matucci-Cerinic M, Romano E. Decreased Serum Levels of SIRT1 and SIRT3 Correlate with Severity of Skin and Lung Fibrosis and Peripheral Microvasculopathy in Systemic Sclerosis. J Clin Med. 2022;11(5):1362.
Brito-Zerón P, Baldini C, Bootsma H, Bowman SJ, Jonsson R, Mariette X, et al. Sjögren syndrome. Nat Rev Dis Primers. 2016;2(1):16047.
Mihai A, Caruntu C, Jurcut C, Blajut FC, Casian M, Opris-Belinski D, et al. The Spectrum of Extraglandular Manifestations in Primary Sjögren’s Syndrome. J Pers Med. 2023;13(6):961.
Kurosawa M, Shikama Y, Furukawa M, Arakaki R, Ishimaru N, Matsushita K. Chemokines Up-Regulated in Epithelial Cells Control Senescence-Associated T Cell Accumulation in Salivary Glands of Aged and Sjögren’s Syndrome Model Mice. Int J Mol Sci. 2021;22(5):2302.
Smoleńska Ż, Pawłowska J, Zdrojewski Z, Daca A, Bryl E. Increased percentage of CD8+CD28− T cells correlates with clinical activity in primary Sjögren’s syndrome. Cell Immunol. 2012;278(1– 2):143–51.
Fessler J, Fasching P, Raicht A, Hammerl S, Weber J, Lackner A, et al. Lymphopenia in primary Sjögren’s syndrome is associated with premature aging of naïve CD4+ T cells. Rheumatology. 2021;60(2):588–97.
Wang X, Bootsma H, Terpstra J, Vissink A, van der Vegt B, Spijkervet FKL, et al. Progenitor cell niche senescence reflects pathology of the parotid salivary gland in primary Sjögren’s syndrome. Rheumatology. 2020;59(10):3003–13.
Zong Y, Yang Y, Zhao J, Li L, Luo D, Hu J, et al. Characterisation of macrophage infiltration and polarisation based on integrated transcriptomic and histological analyses in Primary Sjögren’s syndrome. Front Immunol. 2023;14:1292146.
Pringle S, Wang X, Verstappen GMPJ, Terpstra JH, Zhang CK, He A, et al. Salivary Gland Stem Cells Age Prematurely in Primary Sjögren’s Syndrome. Arthritis & Rheumatology. 2019;71(1):133–42.
Peng X, Wu Y, Brouwer U, van Vliet T, Wang B, Demaria M, et al. Cellular senescence contributes to radiation-induced hyposalivation by affecting the stem/progenitor cell niche. Cell Death Dis. 2020;11(10):854.
Kawashima M, Kawakita T, Maida Y, Kamoi M, Ogawa Y, Shimmura S, et al. Comparison of telomere length and association with progenitor cell markers in lacrimal gland between Sjögren syndrome and non-Sjögren syndrome dry eye patients. Mol Vis. 2011;17:1397–404.
Lu C, Pi X, Xu W, Qing P, Tang H, Li Y, et al. Clinical significance of T cell receptor repertoire in primary Sjogren’s syndrome. EBioMedicine. 2022;84:104252.
Zheng Q, Huang J, Wang G. Mitochondria, Telomeres and Telomerase Subunits. Front Cell Dev Biol. 2019;7.
Javorova P, Fessler J, Rammerstorfer C, Zeiler M, Muralikrishnan AS, Lackner A, et al. POS0294 MITOCHONDRIAL DYSFUNCTION AND SENESCENCE OF NAÏVE T CELLS IN SJÖGREN´S SYNDROME. In: Scientific Abstracts. BMJ Publishing Group Ltd and European League Against Rheumatism; 2023. p. 389.1-389.
Yoon J, Lee M, Ali AA, Oh YR, Choi YS, Kim S, et al. Mitochondrial double-stranded RNAs as a pivotal mediator in the pathogenesis of Sjӧgren’s syndrome. Mol Ther Nucleic Acids. 2022;30:257–69.
Shi H, Zheng L, Zhang P, Yu C. miR-146a and miR- 155 expression in <scp>PBMC</scp> s from patients with Sjögren’s syndrome. Journal of Oral Pathology & Medicine. 2014;43(10):792–7.
Weyand CM, Goronzy JJ. Mediumand Large-Vessel Vasculitis. New England Journal of Medicine. 2003;349(2):160–9.
Weichhart T. mTOR as Regulator of Lifespan, Aging, and Cellular Senescence: A Mini-Review. Gerontology. 2018;64(2):127–34.
Maciejewski-Duval A, Comarmond C, Leroyer A, Zaidan M, Le Joncour A, Desbois AC, et al. mTOR pathway activation in large vessel vasculitis. J Autoimmun. 2018;94:99–109.
Arriola Apelo SI, Lamming DW. Rapamycin: An InhibiTOR of Aging Emerges From the Soil of Easter Island. J Gerontol A Biol Sci Med Sci. 2016;71(7):841–9.
Ianni A, Kumari P, Tarighi S, Argento FR, Fini E, Emmi G, et al. An Insight into Giant Cell Arteritis Pathogenesis: Evidence for Oxidative Stress and SIRT1 Downregulation. Antioxidants. 2021;10(6):885.
Jiemy WF, van Sleen Y, Graver JC, Pringle S, Brouwer E, van der Geest KSM, et al. Indication of Activated Senescence Pathways in the Temporal Arteries of Patients With Giant Cell Arteritis. Arthritis & Rheumatology. 2023;75(10):1812–8.
Antonio AA, Santos RN, Abariga SA. Tocilizumab for giant cell arteritis. Cochrane Database of Systematic Reviews. 2022;2022(5).
Cid MC, Unizony SH, Blockmans D, Brouwer E, Dagna L, Dasgupta B, et al. Efficacy and safety of mavrilimumab in giant cell arteritis: a phase 2, randomised, double-blind, placebo-controlled trial. Ann Rheum Dis. 2022;81(5):653–61.
Kurata A, Saito A, Hashimoto H, Fujita K, Ohno S ichiro, Kamma H, et al. Difference in immunohistochemical characteristics between Takayasu arteritis and giant cell arteritis: It may be better to distinguish them in the same age. Mod Rheumatol. 2019;29(6):992–1001.
Dejaco C, Duftner C, Al-Massad J, Wagner AD, Park JK, Fessler J, et al. NKG 2 D stimulated T-cell autoreactivity in giant cell arteritis and polymyalgia rheumatica. Ann Rheum Dis. 2013;72(11):1852–9.
Jin K, Wen Z, Wu B, Zhang H, Qiu J, Wang Y, et al. NOTCH-induced rerouting of endosomal trafficking disables regulatory T cells in vasculitis. Journal of Clinical Investigation. 2021;131(1).
Watanabe R, Hashimoto M. Pathogenic role of monocytes/macrophages in large vessel vasculitis. Front Immunol. 2022;13.
Watanabe R, Maeda T, Zhang H, Berry GJ, Zeisbrich M, Brockett R, et al. MMP (Matrix Metalloprotease)-9–Producing Monocytes Enable T Cells to Invade the Vessel Wall and Cause Vasculitis. Circ Res. 2018;123(6):700–15.
Coit P, De Lott LB, Nan B, Elner VM, Sawalha AH. DNA methylation analysis of the temporal artery microenvironment in giant cell arteritis. Ann Rheum Dis. 2016;75(6):1196–202.
Croci S, Zerbini A, Boiardi L, Muratore F, Bisagni A, Nicoli D, et al. MicroRNA markers of inflammation and remodelling in temporal arteries from patients with giant cell arteritis. Ann Rheum Dis. 2016;75(8):1527–33.
Kim C, Hu B, Jadhav RR, Jin J, Zhang H, Cavanagh MM, et al. Activation of miR-21-Regulated Pathways in Immune Aging Selects against Signatures Characteristic of Memory T Cells. Cell Rep. 2018;25(8):2148-2162.e5.
Jennette
JC, Falk RJ, Bacon PA, Basu N, Cid MC, Ferrario F, et al. 2012 Revised
International Chapel Hill Consensus Conference Nomenclature of
Vasculitides. Arthritis Rheum. 2013;65(1):1–11.
Moosig F, Csernok E, Wang G, Gross WL. Costimulatory molecules in Wegener’s granulomatosis (WG): lack of expression of CD28 and preferential up-regulation of its ligands B7-1 (CD80) and B7-2 (CD86) on T cells. Clin Exp Immunol. 2001;114(1):113–8.
Kerstein A, Schüler S, Cabral-Marques O, Fazio J, Häsler R, Müller A, et al. Environmental factor and inflammation-driven alteration of the total peripheral T-cell compartment in granulomatosis with polyangiitis. J Autoimmun. 2017;78:79–91.
Lamprecht P. CD28 negative T cells are enriched in granulomatous lesions of the respiratory tract in Wegener’s granulomatosis. Thorax. 2001;56(10):751–7.
Vogt S, Iking-Konert C, Hug F, Andrassy K, Hänsch GM. Shortening of telomeres: Evidence for replicative senescence of T cells derived from patients with Wegener’s granulomatosis. Kidney Int. 2003;63(6):2144–51.
Eriksson P, Sandell C, Backteman K, Ernerudh J. Expansions of CD4+CD28– and CD8+CD28– T cells in Granulomatosis with Polyangiitis and Microscopic Polyangiitis Are Associated with Cytomegalovirus Infection But Not with Disease Activity. J Rheumatol. 2012;39(9):1840–3.
Anaya JM, Lozada-Martinez ID, Torres I, Shoenfeld Y. Autoimmunity in centenarians. A paradox. J Transl Autoimmun. 2024;8:100237.
Anaya JM, Monsalve DM, Rojas M, Rodríguez Y, Montoya-García N, Mancera-Navarro LM, et al. Latent rheumatic, thyroid and phospholipid autoimmunity in hospitalized patients with COVID-19. J Transl Autoimmun. 2021;4:100091.
Meroni PL, Mari D, Monti D, Coppola R, Capri M, Salvioli S, et al. Anti-beta 2 glycoprotein I antibodies in centenarians. Exp Gerontol. 2004;39(10):1459–65.
Avrameas S, Alexopoulos H, Moutsopoulos HM. Natural Autoantibodies: An Undersugn Hero of the Immune System and Autoimmune Disorders—A Point of View. Front Immunol. 2018;9:1320.
Ahuja SK, Manoharan MS, Lee GC, McKinnon LR, Meunier JA, Steri M, et al. Immune resilience despite inflammatory stress promotes longevity and favorable health outcomes including resistance to infection. Nat Commun. 2023;14(1):3286.
Zhou L, Ge M, Zhang Y, Wu X, Leng M, Gan C, et al. Centenarians Alleviate Inflammaging by Changing the Ratio and Secretory Phenotypes of T Helper 17 and Regulatory T Cells. Front Pharmacol. 2022;13:877709.
Frankowska N, Bryl E, Fulop T, Witkowski JM. Longevity, Centenarians and Modified Cellular Proteodynamics. Int J Mol Sci. 2023;24(3):2888.
Santos-Lozano A, Santamarina A, Pareja-Galeano H, Sanchis-Gomar F, Fiuza-Luces C, Cristi-Montero C, et al. The genetics of exceptional longevity: Insights from centenarians. Maturitas. 2016;90:49–57.