EDITORIAL
Oscar Arrieta¹, Rafael Rosell²,³, Andrés F. Cardona⁴
Thoracic Oncology Unit, Instituto Nacional de Cancerología – INCaN, México City, México
Germans Trias i Pujol Research Institute, Badalona (IGTP), Spain
IOR, Hospital Quiron-Dexeus Barcelona, Spain
Institute of Research, Science and Education / Thoracic Oncology Unit, Luis Carlos Sarmiento Angulo Cancer Treatment and Research Center – CTIC, Bogotá, Colombia
Recibido:
1 de noviembre, 2023
Aceptado:
15 de noviembre, 2023
Correspondencia:
DOI: 10.56050/01205498.2304
Precision oncology, defined as molecular profiling of tumors to identify targetable alterations, is rapidly developing, and has entered the mainstream clinical practice. Genomic testing involves stakeholders workingnin a coordinated fashion to deliver high-quality tissue samples to laboratories, where appropriate next-generation sequencing (NGS) molecular analysis leads to actionable results. Clinicians should be familiar with the types of genomic variants reported by the laboratory and the technology used to determine the effects, including limitations of current testing methodologies and reports. Genomic results are best interpreted with multidisciplinary input to reduce uncertainty in clinical recommendations relating to a documented variant (1).
The goals of precision oncology include the expanded use of molecular profiling through the use of multi-omics analysis, the selection of appropriate tissue for typing (including paraffin-embedded tumor samples, fluids, and organoids), the identification of biomarkers with diagnostic and predictive value during treatment and at the time of resistance, and the ease of interpreting the results to facilitate and improve practice, the inclusion of patients in clinical trials, and impact on the long-term outcomes (2,3). Over the past two decades, several lines of research came together nearly simultaneously to promote the evolution of precision oncology. In 1998, the BCR-ABL rearrangement in chronic myeloid leukemia was successfully targeted by imatinib, leading to dramatic clinical remissions and U.S. Food and Drug Administration approval in 2001 (4). After that, the first draft sequence of the human genome was completed the same year (5), followed by the first cancer genome (6).
Rapid discovery of multiple, nonoverlapping driver mutations and tyrosine kinase inhibitors with clinically effective inhibitory properties in solid tumors like lung cancer and melanoma led to assays of alterations performed by polymerase chain reaction (PCR). Using these biomarkers to drive treatment decisions in solid tumors raised expectations and interest in molecular profiling. Sequencing technology and costs improved rapidly during the early 2000s, particularly with the advent of NGS on formalin-fixed, paraffin-embedded tissue, whereby massive parallel sequencing allows the determination of alterations in many genes through a timely, cost-effective process (1).
The concept that all somatic cells have identical genomes must be replaced by a more dynamic model of an increasingly mosaic genome. The engine behind this is somatic mutagenesis. Somatic mutations are low-frequency events only directly detectable when amplified in a clonal lineage (7,8). Using selectable marker genes in transgenic mice and human tissues, several researchers have conclusively demonstrated the accumulation of mutations during aging in various organs and tissues, including the liver, brain, heart, small intestine, and spleen (9). The rate of this increase and the spectrum of mutations (basepair substitutions, deletions, translocations) differed significantly from organ to organ. Over time, accumulating somatic events essentially turns an aging tissue into a mosaic of cells with different genotypes. Owing to the emergence of powerful new methods for analyzing copy number variation (amplifications and deletions) of DNA segments defined by single nucleotide polymorphisms, deletions were observed in the blood and other tissues of humans and mice (10). Such deletions must find their origin in creating somatic subpopulations of cells due to the expansion of a single, de novo mutation.
There is also a large body of evidence that chromosomal aberrations, which are microscopically detectable, accumulate in the blood lymphocytes of both humans and mice (11). Especially aneuploidy, the loss or gain of whole chromosomes resulting in an abnormal numerical karyotype, appears remarkably high, even in postmitotic tissue (11,12). This extraordinarily high level of genome instability during development is maintained with observations of whole-chromosome aneuploidy, segmental deletions, and duplications in individual blastomeres of cleavage-stage embryos (13). Somatic mutation rates differ among different types of genomic sequences. Indeed, in regions containing repeat elements, such as mini-satellites and micro-satellites, retrotransposons, and telomeres, spontaneous mutations can occur at a much higher rate than in single-copy areas. Mutations at microsatellite loci have been found to occur at rates as high as 1×10−2 per locus in humans and increase with age (14). Telomeres (regions of repetitive DNA protecting chromosome ends from deterioration) significantly shorten with age in mammalian cells and tissues, partly due to the end replication problems (14,15).
Rapid progress in nucleotide sequencing technology now allows the detection of even minute post-zygotic mutational differences between twins’ genomes (13). However, mutations present in less than ~10% of all cells in a tissue cannot be detected by direct analysis but require selectable markers. Somatic mosaicism could only be detected because the mutation occurred early during development and/or aging, eventually comprising a sizable fraction of cells. However, these alterations cover a gigantic mountain of mutations unique to an individual cell. In addition, we need accurate insight into the number of genome variants in the different cells comprising a tissue. Indeed, virtually our entire knowledge of normal and cancer genomes has come from studying cells as a bulky mixture. In principle, one could detect somatic mutations in DNA from large cell populations by sequencing thousands of times across the genome and calling all possible variants of the consensus sequence. However, this would be inefficient and expensive and essentially constrained by the high rate of sequencing errors, which can amount to close to 1% (16,17). A possible solution for this problem is to sequence single cells after whole genome amplification and analyze combinations of mutations in an integrated manner. The latter is essential for assessing if random mutations can adversely affect cellular function.
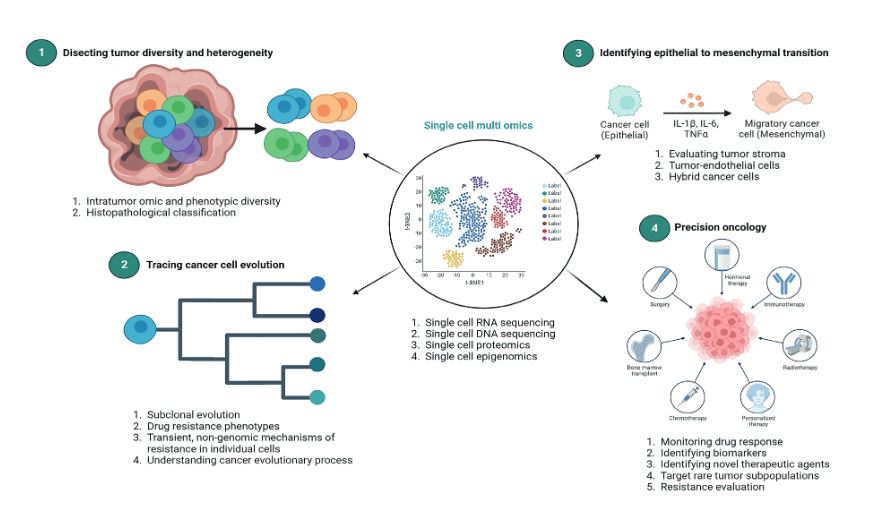
Single-cell omics provide a valuable opportunity to measure different molecules, such as DNA, RNA, protein, and Chromatin, with the highest resolution and technological capacity (18). It is feasible to profile different types of alterations in parallel by simultaneously isolating multiple molecules from a single cell. For example, genomic DNA can be used to assay the single-cell genome, methylome, or chromatin accessibility. In contrast, RNA and proteins from the same cell can be used to profile the transcriptome and proteome, respectively. Using these different single-cell omics profiling strategies, we can design a multi-omics profile for the same cancer cell (19). Currently, we have other technologies to evaluate single-cell multi-omics approaches, such as scG/T-seq (single cell Genome and Transcriptome sequencing), scMT-seq (single cell Methylome and Transcriptome sequencing), scM/T-seq (single cell Methylome and Transcriptome sequencing), scTrio-seq (single-cell triple omics sequencing), and scCOOL-seq (single cell Chromatin Overall Omic-scale Landscape Sequencing) (20). Figure 1 resumes the applications and future of single-cell approaches in cancer precision medicine.
The first single-cell transcriptome analysis was reported in 2009 (21), after which the development of new technologies has been exponential. Recently, experimental protocols that simultaneously sequenced the genome and transcriptome were developed by integrating existing single-cell sequencing methods, namely DR-seq (gDNA and mRNA sequencing) and G/T-seq (Genome and Transcriptome sequencing) (20). In DR-seq, a cell is lysed completely, releasing its DNA and RNA into the same reaction system. Genomic DNA and cDNA are initially homogeneously amplified and distributed for RNA-seq using the CEL-seq protocol and the other for genome sequencing using MALBAC (22,23).
Figure 1. Single-cell approaches in cancer precision medicine.
Unlike DR-seq, G&T- seq separated poly-A-tailed mRNAs from DNA using oligo-dT-coated magnetic beads. Separated mRNA and DNA were sequenced using SMART- seq2 and various WGA protocols (MDA or Pico- PLEX), respectively (23). Most recently, another group reported a novel method for simultaneously isolating genomic DNA and total RNA (SIDR) from single cells using hypotonic synthesis to preserve nuclear lamina integrity and subsequently capturing the cell lysate using antibody-conjugated magnetic microbeads. They found that copy-number variations positively correlated with the corresponding gene expression levels (24). In summary, using DR-seq, G&T-seq, and SIDR, researchers could directly determine the correlation between large-scale copy number variation and transcription levels in the CNV regions.
DNA methylation has been demonstrated to have critical regulatory functions on gene expression in many biological processes. Hence, the DNA methylome and transcriptome relationship from the same single cell is fascinating. Two primary methods for single-cell methylome analysis, single-cell reduced representative bisulfite sequencing (scRRBS) and single-cell whole genome bisulfite sequencing (scWGBS) (25), were previously described. The simultaneous profiling of the methylome and transcriptome of a single cell provides a unique opportunity to directly measure DNA methylation and gene transcription within the same single cell and to study the correlation of DNA methylation differences with gene transcription variance across single cells. For example, scM&T-seq investigated the relationship between the transcriptome and DNA methylome and found that low methylated regions showed high variance in methylation level, consistent with their role as distal regulatory elements that control gene expression (26).
Single-cell sequencing technologies for genome-wide profiling and the subsequent integrative computational analysis methods are central to interpreting single-cell multi-omics data. Considering single-cell genome sequencing, integrating massive amounts of information allows for identifying copy number variation and point mutations/SNPs. Both have been addressed in bulk Wide Genome Sequencing (WGS), and the methods developed for bulk WGS data have guided single-cell WGS analysis. However, the evaluation of knowledge has allowed us to identify the copy number variation using the Hidden Markov Model (HMM) or Circular Binary Segmentation (CBS) (27). Although these two novel methods perform similarly in many situations, user-defined parameter adjustments within the algorithms can affect the sensitivity and specificity of copy number calls. On the other hand, single-cell RNA-seq data enables the discovery of exciting and new biological phenomena while presenting new challenges for analysis. This allows single-cell RNA-seq to identify cell subtypes with unprecedented resolution and reconstruct continuous cell lineages. In addition, some early studies showed that the identification of cell subtypes or reconstruction of cell lineage could be made manually by experts (28). More recently, massive datasets with highly heterogeneous cell populations have precluded the feasibility of manual annotation, and many computational pipelines have been developed, including SINCERA, pcaReduce, SC3, and SNN-Cliq (28). Finally, single-cell methylome analysis allows methods to aggregate methylation levels from adjacent CpG sites or regions with similar biological properties to overcome the sparseness of single-cell genome-wide bisulfite sequencing (scWGBS) data.
The applications of single cell multiomics analysis in cancer include identifying cell subtypes from a heterogeneous cell population. Second, it can be used to reconstruct cell lineage trajectories and understand developmental biology (29). DNA mutations and epigenetic modifications gained during the cell division can be used for lineage tracing. In contrast, the transcriptome of the matching single cells can reveal the concomitant alteration of gene expression and transcriptional cell fate change during cell proliferation and differentiation. Single-cell multi-omics can not only help research determine the occurrence order of different mutations during cancer evolution (29). However, they can also reveal their functional consequences, such as alteration in gene expression, which will eventually help us identify the causal mutations that induce the transition from normal cells to cancer cells. We are rapidly transitioning from in-depth genomic and RNA-seq analysis to multi-omics integration that allows us to segment the evolution of cancer, favor the study of resistance, and recognize new molecular targets potentially valuable for characterizing the disease’s prognosis and predicting the outcomes. Twenty years after the human genome project, we can see the inner universe of humanity and the limit of evolutionary possibilities of solid and hematological tumors. This “Revista Medicina” issue includes definitions, perspectives, and new interventions and technologies in molecular biology.
Schwartzberg L, Kim ES, Liu D, Schrag D. Precision Oncology: Who, How, What, When, and When Not? Am Soc Clin Oncol Educ Book. 2017;37:160-169. doi: 10.1200/EDBK_174176.
Takeuchi S, Okuda S. Knowledge base toward understanding actionable alterations and realizing precision oncology. Int J Clin Oncol. 2019;24(2):123-130. doi: 10.1007/s10147-018-1378-0.
Paolillo C, Londin E, Fortina P. Next generation sequencing in cancer: opportunities and challenges for precision cancer medicine. Scand J Clin Lab Invest Suppl. 2016;245:S84-91. doi: 10.1080/00365513.2016.1210331.
Bumbea H, Vladareanu AM, Voican I, Cisleanu D, Barsan L, Onisai M. Chronic myeloid leukemia therapy in the era of tyrosine kinase inhibitors--the first molecular targeted treatment. J Med Life. 2010;3(2):162-6.
Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, Sutton GG, et al. The sequence of the human genome. Science. 2001;291(5507):1304-51. doi: 10.1126/ science.1058040.
Sjöblom T, Jones S, Wood LD, Parsons DW, Lin J, Barber TD, et al. The consensus coding sequences of human breast and colorectal cancers. Science. 2006;314(5797):268-74. doi: 10.1126/science.1133427.
Piotrowski A, Bruder CE, Andersson R, Diaz de Ståhl T, Menzel U, et al. Somatic mosaicism for copy number variation in differentiated human tissues. Hum Mutat. 2008;29(9):1118-24. doi: 10.1002/humu.20815.
Dollé ME, Snyder WK, Gossen JA, Lohman PH, Vijg J. Distinct spectra of somatic mutations accumulated with age in mouse heart and small intestine. Proc Natl Acad Sci U S A. 2000;97(15):8403-8. doi: 10.1073/ pnas.97.15.8403.
Jacobs KB, Yeager M, Zhou W, Wacholder S, Wang Z, Rodriguez-Santiago B, et al. Detectable clonal mosaicism and its relationship to aging and cancer. Nat Genet. 2012;44(6):651-8. doi: 10.1038/ng.2270.
Frey LJ, Piccolo SR, Edgerton ME. Multiplicity: an organizing principle for cancers and somatic mutations. BMC Med Genomics. 2011;4:52. doi: 10.1186/1755-8794-4-52.
Faggioli F, Wang T, Vijg J, Montagna C. Chromosome-specific accumulation of aneuploidy in the aging mouse brain. Hum Mol Genet. 2012;21(24):5246-53. doi: 10.1093/hmg/dds375.
Zhang L, Vijg J. Somatic Mutagenesis in Mammals and Its Implications for Human Disease and Aging. Annu Rev Genet. 2018;52:397-419. doi: 10.1146/annurev-genet-120417-031501.
Cinnioglu C, Kayali R, Darvin T, Akinwole A, Jakubowska M, Harton G. Aneuploidy Screening using Next Generation Sequencing. Methods Mol Biol. 2019;1885:85-102. doi: 10.1007/978-1-4939-8889- 1_6.
Gallon R, Mühlegger B, Wenzel SS, Sheth H, Hayes C, Aretz S, et al. A sensitive and scalable microsatellite instability assay to diagnose constitutional mismatch repair deficiency by sequencing of peripheral blood leukocytes. Hum Mutat. 2019;40(5):649-655. doi: 10.1002/humu.23721.
Kumar N, Sethi G. Telomerase and hallmarks of cancer: An intricate interplay governing cancer cell evolution. Cancer Lett. 2023:216459. doi: 10.1016/j. canlet.2023.216459.
Salk JJ, Fox EJ, Loeb LA. Mutational heterogeneity in human cancers: origin and consequences. Annu Rev Pathol. 2010;5:51-75. doi: 10.1146/annurev-pathol-121808-102113.
Helleday T, Eshtad S, Nik-Zainal S. Mechanisms underlying mutational signatures in human cancers. Nat Rev Genet. 2014;15(9):585-98. doi: 10.1038/nrg3729.
Zhu Z, Jiang L, Ding X. Advancing Breast Cancer Heterogeneity Analysis: Insights from Genomics, Transcriptomics and Proteomics at Bulk and Single-Cell Levels. Cancers (Basel). 2023;15(16):4164. doi: 10.3390/cancers15164164.
Hu Y, An Q, Sheu K, Trejo B, Fan S, Guo Y. Single Cell Multi-Omics Technology: Methodology and Application. Front Cell Dev Biol. 2018;6:28. doi: 10.3389/ fcell.2018.00028.
Chen C, Wang J, Pan D, Wang X, Xu Y, Yan J, et al. Applications of multi-omics analysis in human diseases. MedComm (2020). 2023;4(4):e315. doi: 10.1002/ mco2.315.
Tang F, Barbacioru C, Wang Y, Nordman E, Lee C, Xu N, et al. mRNA-Seq whole-transcriptome analysis of a single cell. Nat Methods. 2009;6(5):377-82. doi: 10.1038/nmeth.1315.
Dey SS, Kester L, Spanjaard B, Bienko M, van Oudenaarden A. Integrated genome and transcriptome sequencing of the same cell. Nat Biotechnol. 2015;33(3):285-289. doi: 10.1038/nbt.3129.
Macaulay IC, Haerty W, Kumar P, Li YI, Hu TX, Teng MJ, et al. G&T-seq: parallel sequencing of single-cell genomes and transcriptomes. Nat Methods. 2015;12(6):519-22. doi: 10.1038/nmeth.3370.
Han KY, Kim KT, Joung JG, Son DS, Kim YJ, Jo A, et al. SIDR: simultaneous isolation and parallel sequencing of genomic DNA and total RNA from single cells. Genome Res. 2018;28(1):75-87. doi: 10.1101/ gr.223263.117.
Angermueller C, Clark SJ, Lee HJ, Macaulay IC, Teng MJ, Hu TX, et al. Parallel single-cell sequencing links transcriptional and epigenetic heterogeneity. Nat Methods. 2016;13(3):229-232. doi: 10.1038/ nmeth.3728.
Hu Y, Huang K, An Q, Du G, Hu G, Xue J, et al. Simultaneous profiling of transcriptome and DNA methylome from a single cell. Genome Biol. 2016;17:88. doi: 10.1186/s13059-016-0950-z.
Farlik M, Halbritter F, Müller F, Choudry FA, Ebert P, Klughammer J, et al. DNA Methylation Dynamics of Human Hematopoietic Stem Cell Differentiation. Cell Stem Cell. 2016;19(6):808-822. doi: 10.1016/j.stem.2016.10.019.
An Y, Zhao X, Zhang Z, Xia Z, Yang M, Ma Let al. DNA methylation analysis explores the molecular basis of plasma cell-free DNA fragmentation. Nat Commun. 2023;14(1):287. doi: 10.1038/s41467-023-35959-6.
Pan D, Jia D. Application of Single-Cell Multi-Omics in Dissecting Cancer Cell Plasticity and Tumor Heterogeneity. Front Mol Biosci. 2021;8:757024. doi: 10.3389/fmolb.2021.757024.