Cáncer colorrectal: factores de riesgo, diagnóstico y cribado
..................
María José Moreta ¹
, Joan Llach ¹ ², Leticia Moreira ¹
³
Resumen
El cáncer colorrectal (CCR) representa una de las neoplasias más comunes y una de las principales muertes por cáncer. La alta incidencia y mortalidad del CCR refuerzan la necesidad de profundizar en el conocimiento de los factores y grupos de riesgo, estrategias para el diagnóstico precoz, prevención y cribado, con el objetivo final de mejorar el pronóstico de los pacientes que han desarrollado o tienen mayor riesgo de esta neoplasia.
Este artículo presenta una visión actualizada de todos estos puntos relevantes de manejo en la práctica clínica habitual.
Key words: Cáncer colorrectal; cribado; diagnóstico; prevención.
Colorectal cancer: risk factors, diagnosis and screening
Abstract
Colorectal cancer (CRC) represents one of the most common neoplasms and one of the main cancer deaths. The high incidence and mortality of CRC reinforce the need for in- depth knowledge of risk factors and groups, strategies for early diagnosis, prevention and screening, with the ultimate goal of improving the prognosis of patients who have developed or are at increased risk of this neoplasm.
In this review, a brief updated view of all these relevant management points in routine clinical practice will be presented.
Key words: Colorectal cancer; screening; diagnosis; prevention.
..............
² ORCID_IDs: Joan Llach: 0000-0001-6535-8925
³ ORCID_IDs: Leticia Moreira: 0000-0002-4518-8591.
Introducción
Los datos sobre incidencia y mortalidad del cáncer colorrectal (CCR) varían de forma marcada alrededor del mundo. De acuerdo con la base de datos de la WHO-GLOBOCAN del 2020 (1) el CCR es el tercer cáncer más diagnosticado en hombres, después del cáncer de pulmón y del cáncer de próstata, con una incidencia ajustada por edad de 23,4 por 100.000 habitantes, y el segundo en mujeres después del cáncer de mama, con una incidencia ajustada por edad de 16,2 por 100.000 habitantes. En Colombia, en el año 2020 se produjeron 10.783 nuevos casos de CCR, de los cuales 4.960 fueron en hombres y 5.823 en mujeres. Una incidencia que se incrementa de forma importante con la edad y que se duplica a partir de los 50 años (917 casos entre 40 y 49 años versus 2.023 casos entre 50 y 59 años).En cuanto a la mortalidad, el CCR ocupa el tercer lugar, después del cáncer de pulmón y de mama, con una tasa de mortalidad global ajustada por edad de 9 por 100.000 habitantes, la mayor proporción se encuentra en Europa, Asia y Oceanía en comparación con Norteamérica, América Latina y el Caribe. Específicamente en Colombia, la mortalidad ajustada por edad es de 8,2 por 100.000 habitantes, siendo mayor en hombres que en mujeres, con una tasa de mortalidad ajustada por edad de 8,5 por 100.000 y 7,9 por 100.000, respectivamente.
La alta incidencia y mortalidad del CCR refuerzan la necesidad de conocer en profundidad los factores y grupos de riesgo, las estrategias para el diagnóstico precoz, prevención y cribado, con el objetivo final de mejorar el pronóstico de los pacientes que han desarrollado o tienen mayor riesgo de esta neoplasia.
Se presenta una visión actualizada de estos puntos relevantes de manejo en la práctica clínica habitual.
Factores de riesgo del CCR
Factores ambientales
La gran mayoría de los CCRs corresponde a formas esporádicas, que acontecen habitualmente en población de riesgo medio (varones y mujeres mayores de 50 años, sin antecedentes personales ni familiares de esta neoplasia). Los principales factores de riesgo medioambientales asociados al CCR son la edad avanzada, el índice de masa corporal, la Diabetes Mellitus, el sedentarismo y el tipo de dieta. Respecto a este último factor, el consumo elevado de grasa total, colesterol, y carne (roja o blanca), y la dieta baja en calcio y ácido fólico se asocian con el incremento de la incidencia del CCR. Por otro lado, una dieta rica en fibra, frutas, hortalizas y legumbres parece ejercer un papel protector en el desarrollo de dicha neoplasia (2) (3).El tabaquismo y el consumo de alcohol son factores de riesgo bien establecidos (4) (5). Se ha descrito un aumento del CCR entre el 57 y 71 % en grandes fumadores (>1 paquete al día durante un período de exposición prolongado), así como un riesgo significativamente mayor de presentar adenomas y recurrencia del adenoma después de la polipectomía en estos individuos (6). Respecto al alcoholismo, en un metaanálisis de ocho estudios de cohortes, el riesgo relativo (RR) para el consumo de 45 g/día (es decir, alrededor de cuatro bebidas estándar por día) en comparación con los no bebedores fue de 1,41 (intervalo de confianza [IC] del 95 %, 1,16 - 1,72) (7).
Predisposición genética y familiar al CCR
En un porcentaje limitado (<5 %), el CCR se desarrolla en el contexto de formas hereditarias (fundamentalmente poliposis colorrectales y síndrome de Lynch), cuya causa genética está bien establecida, mientras que una proporción aún menor (<1 %) está constituida por tumores que complican una enfermedad inflamatoria intestinal de larga evolución. Por otro lado, en un 25 a 30 % de pacientes con CCR, existen antecedentes familiares de esta neoplasia, aunque sin llegar a cumplir los criterios diagnósticos de las formas hereditarias, denominándose este grupo CCR familiar para distinguirlo de estas últimas (8) (figura 1).Formas hereditarias de CCR
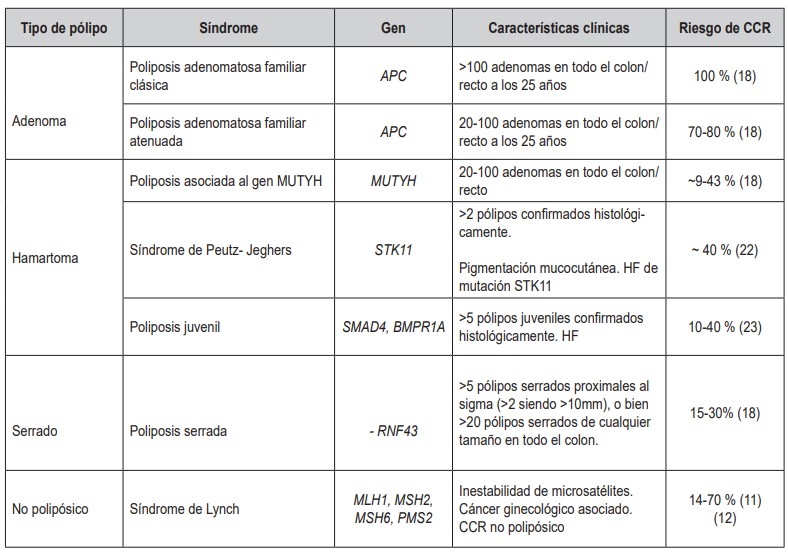
En este grupo se encuentran los individuos con CCR con una mutación germinal conocida asociada a este tumor, y representa entre el 3 y 5 % del total de CCRs. Lo conforman el síndrome de Lynch (CCR hereditario no polipósico) y los síndromes de poliposis con riesgo aumentado de CCR (poliposis adenomatosa familiar, poliposis serrada y poliposis hamartomatosas) (Tabla 1, figura 1).a. Síndrome de Lynch (SL)
El SL es un síndrome hereditario autosómico dominante, que se define por la presencia de variantes patogénicas en línea germinal en los genes de reparación del ADN (MLH1, MSH2, MSH6, PMS2 y deleciones en EPCAM, un regulador 5 de MSH2). Es la causa hereditaria más común de CCR (riesgo hasta del 70 % a lo largo de la vida según el tipo de mutación, y supone alrededor del 3-4 % del total de los CCRs) y de cáncer de endometrio. Se caracteriza por el desarrollo de CCR a edades más tempranas, presencia de cáncer sincrónico y metacrónico, y carcinogénesis acelerada. Además, se asocia con un mayor riesgo de tumores predominantemente epiteliales en otras localizaciones como de ovario, estómago, intestino delgado, páncreas, uréter y pelvis renal, así como cáncer de piel y tumores del sistema nervioso central (9) (10).
El riesgo global de cada tipo de cáncer es específico y varía según el gen afecto. Por ejemplo, el riesgo de CCR a lo largo de la vida varía alrededor del 14 % para los portadores de la variante PMS2 y del 70 % para los portadores de la variante MLH1 (11) (12).
Un correcto diagnóstico del SL facilita las medidas preventivas encaminadas a reducir la incidencia y mortalidad por CCR con programas de vigilancia adaptados y tratamientos específicos (13), sin embargo, supone un reto en la práctica clínica y un elevado grado de sospecha. Actualmente se debería realizar cribado universal de SL a todos los CCR mediante inmunohistoquímica (IHQ) de las proteínas del sistema de reparación del ADN y/o estudio de inestabilidad de microsatélites (IMS), y en caso de identificar alteración en estos análisis, realizar un estudio genético para confirmar que se trata de una mutación germinal en alguno de los genes correspondientes (14).
b. Poliposis adenomatosa familiar (PAF)
La PAF es una forma hereditaria de CCR cuya causa suele ser una mutación germinal en el gen APC o MUTYH. Esta última se conoce también como poliposis asociada a MUTYH y presenta un patrón de herencia autosómica recesiva, a diferencia del resto de poliposis (15).
La PAF se caracteriza por el desarrollo de múltiples adenomas en el colon y recto (>100 en su variante clásica y >10-20 en su variante atenuada) con un riesgo elevado variable de CCR, siendo prácticamente del 100 % en su forma clásica antes de los 40 años. En la PAF atenuada la aparición de adenomas se retrasa, con una edad media de presentación a los 44 años, son menos numerosos y se localizan predominantemente en el colon derecho. En las formas atenuadas, la probabilidad de CCR disminuye, aunque sigue siendo alta, con una incidencia acumulada del 70 % a los 70 años (16).
La manifestación extracolónica más frecuente es la aparición de pólipos en el tracto digestivo superior. Los pólipos gástricos son frecuentes, pero el 90 % de ellos son pólipos de glándulas fúndicas de naturaleza benigna. No obstante, el duodeno supone la segunda localización más frecuente sobre la que asientan los adenomas asociados a la PAF, con un riesgo acumulado de casi el 100 % a los 70 años en las formas clásicas. Normalmente los pólipos se localizan en la segunda y tercera porción duodenal, y el riesgo de cáncer duodenal está entre el 4 y12 % a lo largo de la vida.
La PAF también se asocia con otras neoplasias como el adenocarcinoma de páncreas, el hepatoblastoma, el cáncer papilar de tiroides o tumores cerebrales, con manifestaciones benignas como la hipertrofia congénita del epitelio de la retina, los quistes epidermoides, los osteomas y los tumores desmoides (17).
c. Síndrome de poliposis serrada (SPS)
Los criterios clínicos de la SPS se actualizaron recientemente; incluyen individuos con cinco o más pólipos serrados proximales al recto >5 mm (al menos dos de ellos con un tamaño de 10 mm o más), o bien personas con 20 o más pólipos serrados en todo el colon con al menos cinco de ellos proximales al recto (18).
Existe un riesgo significativo de cáncer de colon en estos individuos a través de la llamada “vía serrada” de la carcinogénesis, desarrollándose hasta un 30 % del total de los CCR por esta vía (19). Tan solo el 3 % de los casos de SPS se explican por mutaciones identificables de la línea germinal, siendo las mutaciones en RNF43 la única causa genética actualmente validada (20). La prevalencia de SPS se estima entre 1:151 y 1:294 de los pacientes sometidos a una colonoscopia, debida a un test de sangre oculta en heces positivo en programas de cribado poblacional, porcentaje significativamente mayor a otras poliposis como la PAF (21).
Finalmente, existen otras entidades como la poliposis hamartomatosa juvenil (variante patogénica germinal en BMPR1A, SMAD4 o ENG), el síndrome de Peutz Jeghers (STK11) o el síndrome de Cowden (PTEN) que también predisponen al desarrollo de CCR, aunque con una incidencia y riesgo menor que las explicadas anteriormente (Tabla 1, figura 1).
CCR familiar
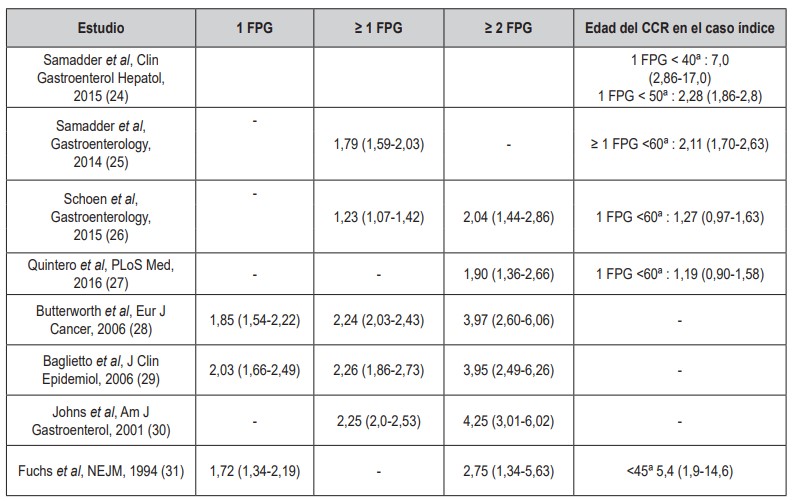
Los individuos con antecedentes familiares presentan un mayor riesgo de desarrollar CCR, probablemente por compartir factores de riesgo tanto ambientales como genéticos no identificados. Se define como CCR familiar cuando existe agregación de esta neoplasia y se ha descartado un SL o síndromes polipósicos. El riesgo de CCR en los familiares de pacientes con esta neoplasia está condicionado por el grado de parentesco, número de familiares afectos y la edad del caso índice. Se ha observado que el riesgo relativo de CCR disminuye en la medida que aumenta la edad al diagnóstico del familiar afecto (24). El riesgo relativo de CCR más elevado se encuentra en los individuos <50 años con un familiar de primer grado (FPG) con CCR <40 años (Riesgo relativo -RR- 7,0 [IC 95 % 2,86 -17,0]) comparado con individuos <50 años sin historia familiar de CCR (Tabla 2).Diagnóstico
Signos y síntomas
El CCR puede ser detectado en pacientes asintomáticos en un estadio precoz como resultado de las técnicas de cribado (32) o en pacientes con síntomas como rectorragia, melenas, dolor abdominal, pérdida de peso, anemia ferropénica, cambio del hábito intestinal, masa abdominal palpable, náuseas, vómitos e incluso complicaciones agudas como obstrucción, perforación o hemorragia, en estadios avanzados cuando el tumor ha crecido hacia la luz o se ha extendido hacia estructuras adyacentes. La extensión del CCR puede producirse por cualquier vía, siendo la más frecuente la vía linfática hacia ganglios regionales, seguida de la vía hematógena, generando implantes en hígado y pulmones, por el sistema de drenaje venoso portal del colon y finalmente por continuidad produciendo implantes peritoneales; en algunos casos, el primer lugar de metástasis son los pulmones, pero esto se produce cuando el tumor se origina en recto distal porque el sistema venoso drena directamente en la vena cava.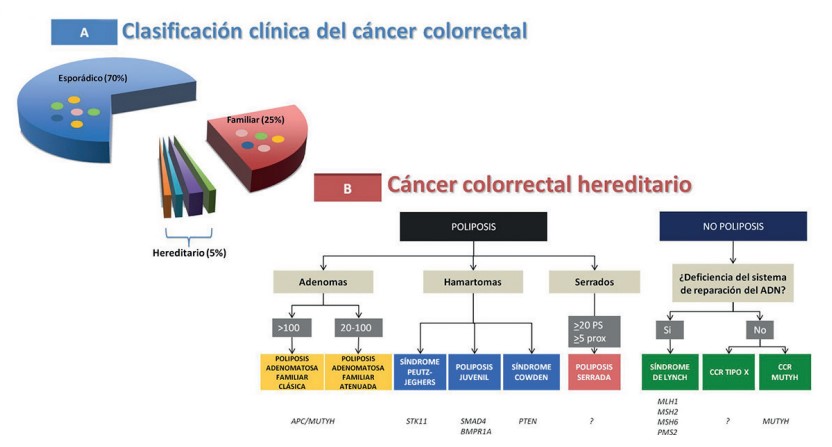
Figura 1. Clasificación del cáncer colorrectal. A) Clasificación clínica: esporádico, familiar, hereridatio. B) Tipos de cáncer colorectal hereditario: polipósico y no polipósico y los genes asociados.
Se describe en varios estudios que la presencia de síntomas influye negativamente en el pronóstico (32) (35). Específicamente, aquellos que no fueron diagnosticados mediante técnicas de cribado tienen mayor riesgo de presentar al momento del diagnóstico un tumor invasivo ≥T3 (RR 1,96), afectación ganglionar (RR 1,92) y enfermedad metastásica (RR 3,37). También asocian mayor riesgo de muerte (RR 3,02) y recurrencia (RR 2,19), así como periodos más cortos de supervivencia e intervalos libres de enfermedad (36). De igual forma, la presencia de complicaciones agudas se asocian con mal pronóstico, independientemente del estadio del cáncer (37). Todo esto ratifica la importancia de insistir en el uso de las técnicas de cribado para la detección precoz del CCR.
Proceso diagnóstico
Una vez que se sospecha la presencia de CCR es importante realizar una historia clínica completa que recoja adecuadamente sus antecedentes y el proceso actual, además de obtener una muestra de tejido a través de una biopsia endoscópica o de una pieza quirúrgica que permita realizar el diagnóstico histológico definitivo del tumor primario.Tabla 1. Síndromes asociados con un mayor riesgo de cáncer colorrectal. HF: Historia familiar; CCR: Cáncer colorrectal
Siempre que no existan complicaciones que impidan la tolerancia a la preparación intestinal o que requieran tratamiento quirúrgico, como obstrucción, perforación o hemorragia, se debe realizar una colonoscopia. El estudio endoscópico es el estándar de oro para diagnosticar el CCR, ya que permite obtener una biopsia de tejido, localizar la lesión, identificar la presencia de lesiones sincrónicas y extraer pólipos (38). Incluso, en ocasiones las lesiones se pueden extraer completamente y en caso de que se sospeche invasión de la submucosa (por ejemplo, pólipos sésiles o planos de >10 mm), se aconseja marcar la zona con un tatuaje de 3 cm distal a la lesión, para poder localizarla en caso de identificar un cáncer invasivo y realizar una resección local (39).
Hay que tener en cuenta que hasta un 12 % de las colonoscopias no se completan de forma exitosa y es en estos casos en los que la colonografía por TC o también llamada colonoscopia virtual resulta una alternativa útil. Esta técnica brinda una perspectiva endoluminal simulada por computadora del colon distendido por aire y requiere una preparación con dieta líquida de 24 h y limpieza intestinal con laxante, ya que cualquier resto fecal puede simular un pólipo. La evidencia actual muestra que tanto esta técnica como la colonoscopia tienen una sensibilidad similar para detectar CCR y pólipos grandes (40). A pesar de esta evidencia, las posibilidades diagnósticas y terapéuticas que ofrece la colonoscopia, la ubican como el estándar de oro para la investigación de los síntomas sugestivos de CCR (38).
Tabla 2. Riesgo Relativo (RR) de cáncer colorrectal (CCR) con base en el número de familiares de primer grado (FPG) afectos. Se pueden observar varios estudios en los que se evalúa el riesgo relativo de CCR según la historia familiar y edad del diagnóstico del caso índice, objetivándose que a mayor número de FPG afectos y a edades inferiores, el RR aumenta considerablemente.
Parte del proceso diagnóstico debe incluir una analítica completa con hemograma, coagulación, pruebas de función renal, hepática y un perfil nutricional que permitan identificar el estado basal del paciente. La determinación de niveles séricos de marcadores tumorales asociados al CCR como el antígeno carcinoembrionario (CEA) o el antígeno para carbohidrato 19-9 (CA 19-9) no está justificada por su baja sensibilidad y especificidad para el cribado y el diagnóstico (41). Sin embargo, los niveles de CEA resultan útiles en el seguimiento y determinación del pronóstico. Para ello se deben establecer los niveles después de una resección quirúrgica y monitorizarlos; característicamente los niveles altos (>5 ng/ml) confieren peor pronóstico al asociar mayor riesgo de recurrencia durante el primer año. En cuanto al seguimiento, los niveles que no se normalicen tras la cirugía han de sugerir enfermedad residual, y si a largo plazo se identifica un incremento de los mismos se debería sospechar la presencia de una enfermedad metastásica (42).
Estadiaje
Establecido el diagnóstico histopatológico, es necesario determinar la extensión local y a distancia del tumor para definir el tratamiento y el pronóstico. El estadiaje es clínico y se inicia con la exploración física, prestando especial atención a la presencia de ascitis, hepatomegalia, linfadenopatías y la realización de examen rectal digital. A continuación se añaden estudios complementarios:Tomografía computarizada (TC): se planteará la realización de una TC de tórax, abdomen y pelvis para determinar la extensión regional, metástasis a ganglios linfáticos y a distancia, además de posibles complicaciones (obstrucción, perforación o fístulas) (43-44). Sin embargo, la realización de TC torácica es controvertida, porque entre el 10 y 30 % de los casos se encuentran lesiones torácicas indeterminadas que complican el proceso diagnóstico. No obstante, el riesgo de malignidad de estas lesiones es menor al 1 % por lo que justificaría su realización, sobre todo en cáncer de recto ya que el drenaje venoso de las venas hemorroidales hacia la vena cava hace que sea un lugar frecuente de metástasis a distancia (45).
Sigmoidoscopia rígida, ecografía transrectal, ecografía endoscópica transrectal o resonancia magnética pélvica son estudios que en cáncer de recto ayudan a precisar la localización del tumor y afectación del esfínter anal para determinar la posibilidad de resección local, radical o necesidad de terapia preoperatoria (46).
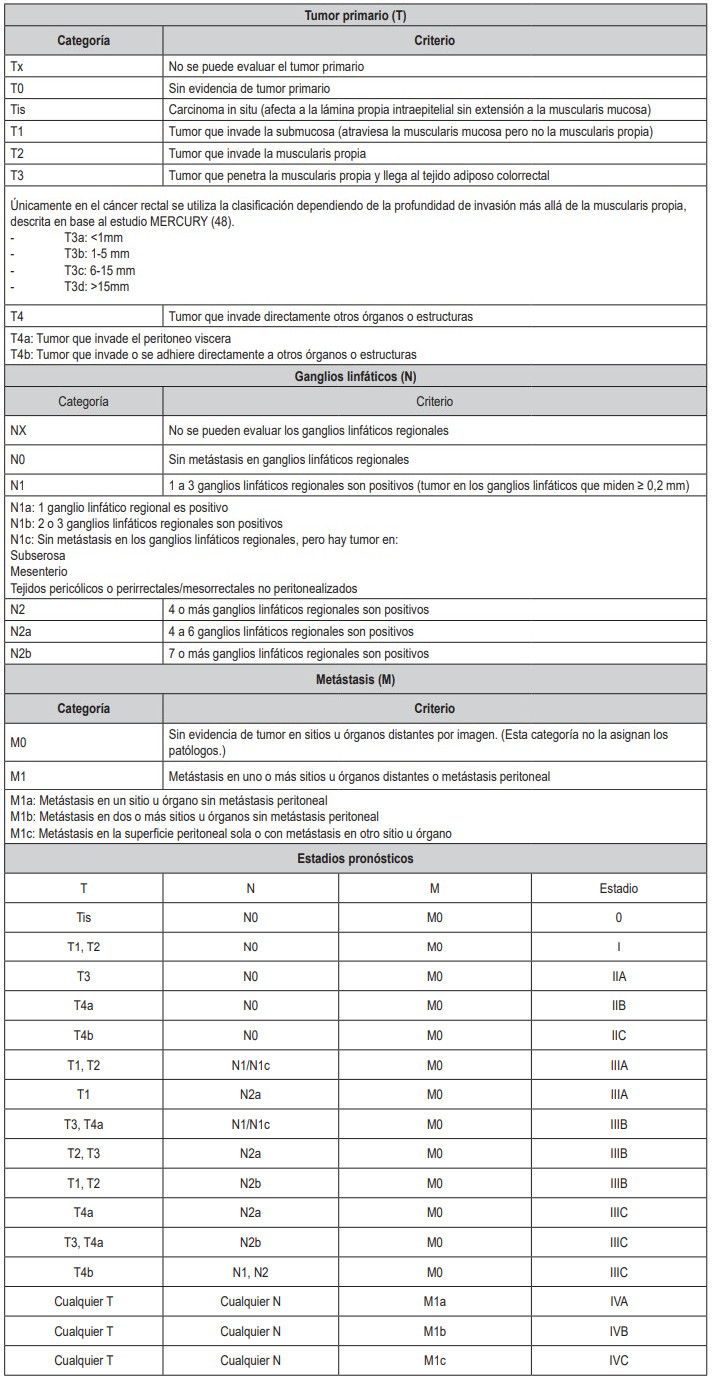
Para estadificar clínicamente el CCR, tras obtener el resultado de las pruebas de imagen, se utiliza el sistema de estadificación Tumor, Nódulo, Metástasis (TNM) cuya revisión más reciente es la octava edición del año 2017 (Tabla 3), junto con los estadios pronósticos asociados (47).
Tratamiento
El tratamiento depende del estadio, pudiendo ser endoscópico, quirúrgico (asociado o no a tratamiento con quimioterapia/radioterapia) o paliativo. El abordaje terapéutico no es objetivo de la revisión actual.Cribado
El conocimiento de la historia natural y de los factores patogénicos implicados en el CCR ha permitido instaurar programas preventivos para evitar su aparición (prevención primaria), detectarlo de una forma precoz (prevención secundaria) y mejorar su pronóstico en el momento del diagnóstico (prevención terciaria). En el caso del CCR, la prevención secundaria se basa en las estrategias de cribado mediante test de sangre oculta en heces (TSOH) y/o colonoscopia /sigmoidoscopia, y tiene como objetivo identificar sujetos asintomáticos con lesiones precancerosas o en una fase inicial de la progresión tumoral.Una buena estrategia de cribado, a nivel general y específicamente en el caso del CCR, debe ser sensible a la detección de lesiones precursoras, debe ser específica, costo/efectiva, cambiar la historia natural de la enfermedad y ser aceptada por la población a la que se va a realizar (49)
Existen distintos escenarios en los cuales la estrategia de cribado va a ser diferente (figura 2).
Cribado de CCR en la población general
En este grupo se incluyen los individuos que presentan un riesgo medio de padecer CCR, es decir, la población >50 años sin antecedentes familiares ni condiciones genéticas conocidas que conlleven un riesgo aumentado de CCR. En la mayoría de los países es la detección de sangre oculta en heces la forma más habitual de cribado en este grupo. No obstante, la sigmoidoscopia directa cada 5 años o la colonoscopia cada 10 años, son estrategias de cribado igualmente validadas.Tabla 3. Estadificación del cáncer colorrectal. Sistema Tumor, Nódulo, Metástasis (TNM), junto con los estadios pronósticos asociados
El test de sangre oculta en heces basado en la determinación de la hemoglobina humana mediante anticuerpos específicos (SOHi) es una técnica que presenta ventajas respecto a otros test fecales como el del guayaco, ya que tiene una mayor sensibilidad y requiere únicamente una muestra de heces (50). Es una prueba que cumple con los principios de una buena estrategia de cribado y por ello en la población general parece la técnica de elección de entrada (51). Se debe realizar con periodicidad anual o bienal hasta los 80 años, y aquellos individuos que presenten un SOHi patológico se deberán someter a una colonoscopia.
Para este grupo, el cribado del CCR con detección de SOH mediante el test inmunológico es efectivo y disminuye la mortalidad específica por CCR (52) (53).
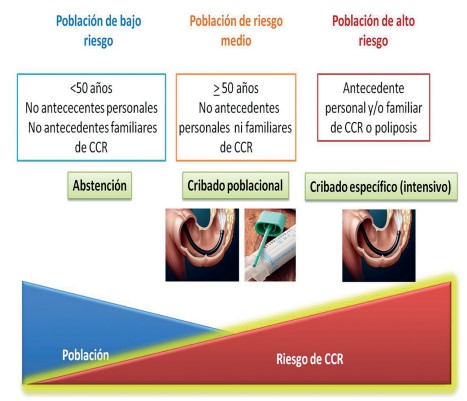
Figura 2. Grupos de riesgo de cáncer colorrectal y cribado recomendado en cada grupo.
Cribado de CCR en individuos con SL
La colonoscopia como estrategia directa ha demostrado reducir la incidencia y mortalidad asociada con CCR en el síndrome de Lynch. Estas pruebas deberían realizarse por endoscopistas especializados con endoscopios de alta definición. Se recomienda iniciar el cribado a los 25 años en individuos con variantes en MLH1 y MSH2, y a los 35 años en MSH6 y PMS2, o bien 5 años antes de la edad de diagnóstico del CCR en el familiar afecto más joven, con una periodicidad bianual (18).
Cribado de CCR en individuos con poliposis adenomatosa familiar y síndrome de poliposis serrada
En ambos casos deberá valorarse la realización de colonoscopias de vigilancia cada 1-2 años, realizando resección de todos los pólipos siempre que sea posible (o en su defecto, de aquellos >3-5 mm). El cribado deberá realizarse a edades tempranas: desde el diagnóstico en la poliposis serrada y antes de los 20 años en la PAF, individualizándose según el riesgo y el fenotipo del síndrome. Si los pólipos no son resecables por tamaño o número, deberá considerarse la realización de una colectomía total con anastomosis ileorrectal (o en su defecto una proctocolectomía total), y tras la cirugía se deberán realizar rectoscopias de control con periodicidad anual o bienal (18).Cribado de CCR en individuos sanos con antecedentes familiares (AF) de dicha neoplasia
Tal y como hemos mencionado anteriormente, los individuos con AF de CCR tienen un riesgo significativamente mayor de este cáncer a lo largo de su vida.Sin embargo, el riesgo difiere considerablemente en función del número de familiares y de la edad al momento del diagnóstico del familiar o familiares afectos (Tabla 2).
De este modo, en individuos con un FPG diagnosticado y mayores de 60 años, o bien uno o más familiares de segundo grado (independientemente de la edad), la estrategia de cribado será la misma que en la población general (SOHi anual o bienal a partir de los 50 años) (49).
En los individuos con un AF de CCR de primer grado, <60 años, una estrategia aceptada sería la realización de colonoscopia directa a partir de los 50 años o 10 años antes del caso más joven. Por último, en individuos con >2 AF de primer grado se podría considerar la realización de colonoscopia directa a partir de los 40 años o 10 años antes del caso más joven, cada 5 años.
Nuevas estrategias de cribado y perspectivas de futuro
El cribado actual (con SOHi o endoscopia) ha demostrado ser eficaz y costo-efectivo, reduce la incidencia y mejora los porcentajes de supervivencia a los 5 años (54). Ningún método es ideal, por ejemplo, la SOHi presenta una alta sensibilidad pero baja especificidad, lo que conlleva a la realización de colonoscopias innecesarias (51), por lo que se trabaja arduamente en la identificación de nuevos biomarcadores no invasivos para el diagnóstico precoz del CCR en diferentes tipos de muestras biológicas (sangre, heces, saliva), identificando ADN tumoral, miRNAs, proteínas, metabolitos, microbiota, etc. (55). Los resultados de múltiples estudios en este aspecto son prometedores, aunque está todavía por determinar cuál es el fluido más conveniente y qué biomarcador o panel de biomarcadores sería el más apropiado, de cara a la identificación de los mejores tests no invasivos, sensibles, específicos, costo-efectivos y de fácil implementación para que impacten realmente en la salud pública.Conflictos de interés
Los autores declaran no tener ningún conflicto de intereses.
Referencias
1. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global Cancer Statistics 2020:
GLOBOCAN Estimates of Incidence and Mortality
Worldwide for 36 Cancers in 185 Countries. CA Cancer
J Clin. 2021;71(3):209-49.
2. Bingham SA. Diet and large bowel cancer. J R Soc Med [Internet]. 1990 Jul;83(7):420-2. Available from:
http://www.ncbi.nlm.nih.gov/pubmed/2203904
3. Potter JD. Reconciling the epidemiology, physiology, and molecular biology of colon cancer. JAMA. 1992;268(12):1573-7.
4. Baron JA, Sandler RS, Haile RW, Mandel JS, Mott LA, Greenberg ER. Folate intake, alcohol consumption, cigarette smoking, and risk of colorectal adenomas. J Natl Cancer Inst. 1998 Jan 7;90(1):57-62.
5. Knekt P, Hakama M, Järvinen R, Pukkala E, Heliövaara M. Smoking and risk of colorectal cancer. Br J Cancer. 1998 Jul;78(1):136-9.
6. Neugut AI, Jacobson JS, De Vivo I. Epidemiology of colorectal adenomatous polyps. Cancer Epidemiol Biomarkers Prev. 1993;2(2):159-76.
7. Cho E, Smith-Warner SA, Ritz J, van den Brandt PA, Colditz GA, Folsom AR, et al. Alcohol intake and colorectal cancer: a pooled analysis of 8 cohort studies. Ann Intern Med. 2004;140(8):603-13.
8. Castells A, Castellví-Bel S, Balaguer F. Concepts in familial colorectal cancer: where do we stand and what is the future? Gastroenterology. 2009;137(2):404-9.
9. Møller P, Seppälä TT, Bernstein I, Holinski-Feder E, Sala P, Gareth Evans D, et al. Cancer risk and survival in path_MMR carriers by gene and gender up to 75 years of age: a report from the Prospective Lynch Syndrome Database. Gut. 2018;67(7):1306-16.
10. Jenkins MA, Baglietto L, Dowty JG, Van Vliet CM, Smith L, Mead LJ, et al. Cancer risks for mismatch repair gene mutation carriers: a population-based early onset case-family study. Clin Gastroenterol Hepatol. 2006;4(4):489-98.
11. Järvinen HJ, Aarnio M, Mustonen H, Aktan-Collan K, Aaltonen LA, Peltomäki P, et al. Controlled 15-year trial on screening for colorectal cancer in families with hereditary nonpolyposis colorectal cancer. Gastroenterology. 2000;118(5):829-34.
12. Edelstein DL, Axilbund J, Baxter M, Hylind LM, Romans K, Griffin CA, et al. Rapid development of colorectal neoplasia in patients with Lynch syndrome. Clin Gastroenterol Hepatol. 2011;9(4):340-3.
13. Sánchez A, Roos VH, Navarro M, Pineda M, Caballol B, Moreno L, et al. Quality of Colonoscopy Is Associated With Adenoma Detection and Postcolonoscopy Colorectal Cancer Prevention in Lynch Syndrome. Clin Gastroenterol Hepatol. 2022;20(3):611-621.e9.
14. Moreira L, Balaguer F, Lindor N, de la Chapelle A, Hampel H, Aaltonen LA, et al. Identification of Lynch Syndrome Among Patients With Colorectal Cancer. JAMA. 2012;308(15):1555.
15. Moreira L, Castells A. Surveillance of patients with hereditary gastrointestinal cancer syndromes. Best Pract Res Clin Gastroenterol. 2016;30(6):923-35.
16. Stjepanovic N, Moreira L, Carneiro F, Balaguer F, Cervantes A, Balmaña J, et al. Hereditary gastrointestinal cancers: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2019;30(10):1558-71.
17. Balaguer F. Hereditary and familial colorectal cancer. Gastroenterol Hepatol. 2014;37(Suppl 3):77-84.
18. van Leerdam ME, Roos VH, van Hooft JE, Dekker E, Jover R, Kaminski MF, et al. Endoscopic management of polyposis syndromes: European Society of Gastrointestinal Endoscopy (ESGE) Guideline. Endoscopy. 2019;51(9):877-95.
19. Carballal S, Rodríguez-Alcalde D, Moreira L, Hernández L, Rodríguez L, Rodríguez-Moranta F, et al. Colorectal cancer risk factors in patients with serrated polyposis syndrome: a large multicentre study. Gut. 2016;65(11):1829- 37.
20. Quintana I, Mejías-Luque R, Terradas M, Navarro M, Piñol V, Mur P, et al. Evidence suggests that germline RNF43 mutations are a rare cause of serrated polyposis. Gut. 2018;67(12):2230-2.
21. Moreira L, Pellisé M, Carballal S, Bessa X, Ocaña T, Serradesanferm A, et al. High prevalence of serrated polyposis syndrome in FIT-based colorectal cancer screening programmes. Gut. 2013;62(3):476-7.
22. Giardiello FM, Trimbath JD. Peutz-Jeghers syndrome and management recommendations. Clin Gastroenterol Hepatol. 2006;4(4):408-15. 23. Brosens LAA, van Hattem A, Hylind LM, Iacobuzio-Donahue C, Romans KE, Axilbund J, et al. Risk of colorectal cancer in juvenile polyposis. Gut. 2007;56(7):965-7.
24. Samadder NJ, Smith KR, Hanson H, Pimentel R, Wong J, Boucher K, et al. Increased Risk of Colorectal Cancer Among Family Members of All Ages, Regardless of Age of Index Case at Diagnosis. Clin Gastroenterol Hepatol. 2015;13(13):2305-2311.e2.
25. Samadder NJ, Curtin K, Tuohy TMF, Pappas L, Boucher K, Provenzale D, et al. Characteristics of missed or interval colorectal cancer and patient survival: a populationbased study. Gastroenterology. 2014;146(4):950-60.
26. Schoen RE, Razzak A, Yu KJ, Berndt SI, Firl K, Riley TL, et al. Incidence and mortality of colorectal cancer in individuals with a family history of colorectal cancer. Gastroenterology. 2015;149(6):1438-1445.e1.
27. Quintero E, Carrillo M, Leoz M-L, Cubiella J, Gargallo C, Lanas A, et al. Risk of Advanced Neoplasia in First-Degree Relatives with Colorectal Cancer: A Large Multicenter Cross-Sectional Study. PLoS Med. 2016 May;13(5):e1002008.
28. Butterworth AS, Higgins JPT, Pharoah P. Relative and absolute risk of colorectal cancer for individuals with a family history: a meta-analysis. Eur J Cancer. 2006;42(2):216-27.
29. Baglietto L, Jenkins MA, Severi G, Giles GG, Bishop DT, Boyle P, et al. Measures of familial aggregation depend on definition of family history: meta- analysis for colorectal cancer. J Clin Epidemiol. 2006;59(2):114-24.
30. Johns LE, Houlston RS. A systematic review and metaanalysis of familial colorectal cancer risk. Am J Gastroenterol. 2001;96(10):2992-3003.
31. Fuchs CS, Giovannucci EL, Colditz GA, Hunter DJ, Speizer FE, Willett WC. A prospective study of family history and the risk of colorectal cancer. N Engl J Med. 1994;331(25):1669-74.
32. Moreno CC, Mittal PK, Sullivan PS, Rutherford R, Staley CA, Cardona K, et al. Colorectal Cancer Initial Diagnosis: Screening Colonoscopy, Diagnostic Colonoscopy, or Emergent Surgery, and Tumor Stage and Size at Initial Presentation. Clin Colorectal Cancer. 2016;15(1):67-73.
33. Ford AC, Veldhuyzen Van Zanten SJO, Rodgers CC, Talley NJ, Vakil NB, Moayyedi P. Diagnostic utility of alarm features for colorectal cancer: Systematic review and meta-analysis. Gut. 2008;57(11):1545-52.
34. Shapley M, Mansell G, Jordan JL, Jordan KP. Positive predictive values of ≥5% in primary care for cancer: Systematic review. Br J Gen Pract. 2010;60(578):366-77.
35. McDermott FT, Hughes ESR, Pihl E, Milne BJ, Price AB. Prognosis in relation to symptom duration in colon cancer. Br J Surg. 1981;68(12):846-9.
36. Amri R, Bordeianou LG, Sylla P, Berger DL. Impact of screening colonoscopy on outcomes in colon cancer surgery. JAMA Surg. 2013;148(8):747-54.
37. Setti Carraro PG, Segala M, Cesana BM, Tiberio G. Obstructing colonic cancer: Failure and survival patterns over a ten-year follow-up after one-stage curative surgery. Dis Colon Rectum. 2001;44(2):243-50.
38. Argilés G, Tabernero J, Labianca R, Hochhauser D, Salazar R, Iveson T, et al. Localised colon cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol Off J Eur Soc Med Oncol. 2020;31(10):1291-305.
39. Ferlitsch M, Moss A, Hassan C, Bhandari P, Dumonceau JM, Paspatis G, et al. Colorectal polypectomy and endoscopic mucosal resection (EMR): European Society of Gastrointestinal Endoscopy (ESGE) Clinical Guideline. Endoscopy. 2017;49(3):270-97.
40. Atkin W, Dadswell E, Wooldrage K, Kralj-Hans I, Von Wagner C, Edwards R, et al. Computed tomographic colonography versus colonoscopy for investigation of patients with symptoms suggestive of colorectal cancer (SIGGAR): A multicentre randomised trial. Lancet. 2013;381(9873):1194-202.
41. Liu Z, Zhang Y, Niu Y, Li K, Liu X, Chen H, et al. A systematic review and meta-analysis of diagnostic and prognostic serum biomarkers of colorectal cancer. PLoS One. 2014;9(8).
42. Konishi T, Shimada Y, Hsu M, Tufts L, Jimenez-Rodriguez R, Cercek A, et al. Association of preoperative and postoperative serum carcinoembryonic antigen and colon cancer outcome. JAMA Oncol. 2018;4(3):309-15.
43. Horton K, Abrams R, Fishman E. Spiral CT of colon cancer: imaging features and role in management. Radiographics. 2000;
44. Nerad E, Lahaye MJ, Maas M, Nelemans P, Bakers FCH, Beets GL, et al. Diagnostic Accuracy of CT for Local Staging of Colon Cancer: A Systematic Review and Meta-Analysis. Am J Roentgenol. 2016 Nov;207(5):984- 95.
45. Kirke R, Rajesh A, Verma R, Bankart MJG. Rectal cancer: Incidence of pulmonary metastases on thoracic CT and correlation with T staging. J Comput Assist Tomogr. 2007;31(4):569-71.
46. Burdan F, Sudol-Szopinska I, Staroslawska E, Kolodziejczak M, Klepacz R, Mocarska A, et al. Magnetic resonance imaging and endorectal ultrasound for diagnosis of rectal lesions. Eur J Med Res. 2015 Jan 14;20:4.
47. Tong GJ, Zhang GY, Liu J, Zheng ZZ, Chen Y, Niu PP, et al. Comparison of the eighth version of the American joint committee on cancer manual to the seventh version for colorectal cancer: A retrospective review of our data. World J Clin Oncol. 2018;9(7):148-61.
48. Taylor FGM, Quirke P, Heald RJ, Moran B, Blomqvist L, Swift I, et al. Preoperative high-resolution magnetic resonance imaging can identify good prognosis stage I, II, and III rectal cancer best managed by surgery alone: a prospective, multicenter, European study. Ann Surg. 2011 Apr;253(4):711-9.
49. Amador F, Bellas B, Clofent J, Carballal S, Cubiella J, Ferrándiz J, et al. Guía de práctica clínica de diagnóstico y prevención del cáncer colorrectal. In Asociación Española de Gastroenterología y Sociedad Española de Medicina de Familia y Comunitaria; 2018.
50. Castells A. [Colorectal cancer screening]. Gastroenterol Hepatol. 2015 Sep;38 Suppl 1:64-70.
51. Quintero E, Castells A, Bujanda L, Cubiella J, Salas D, Lanas Á, et al. Colonoscopy versus fecal immunochemical testing in colorectal-cancer screening. N Engl J Med. 2012;366(8):697-706.
52. Pignone M, Saha S, Hoerger T, Mandelblatt J. Costeffectiveness analyses of colorectal cancer screening: a systematic review for the U.S. Preventive Services Task Force. Ann Intern Med. 2002;137(2):96-104.
53. Levin B, Lieberman DA, McFarland B, Smith RA, Brooks D, Andrews KS, et al. Screening and surveillance for the early detection of colorectal cancer and adenomatous polyps, 2008: a joint guideline from the American Cancer Society, the US Multi-Society Task Force on Colorectal Cancer, and the American College of Radiology. CA Cancer J Clin. 58(3):130-60.
54. Carethers JM, Jung BH. Genetics and Genetic Biomarkers in Sporadic Colorectal Cancer. Gastroenterology. 2015 Oct;149(5):1177-1190.e3.
55. Duran-Sanchon S, Herrera-Pariente C, Moreira L. New non-invasive biomarkers for colorectal cancer screening. Rev Española Enfermedades Dig. 2020;112.
Recibido: 16 de Abril de 2022
Aceptado: 08 de Septiembre de 2022
Correspondencia:
Leticia Moreira
lmoreira@clinic.cat
Aceptado: 08 de Septiembre de 2022
Correspondencia:
Leticia Moreira
lmoreira@clinic.cat