CAMINANDO A HOMBROS DE GIGANTES: INTERSECCIÓN ENTRE LA GENÓMICA Y LA IA
..................
Andrés F. Cardona ¹⁻³, Alejandro Ruíz-Patiño ²,³, Elvira Jaller ³,⁴, July Rodríguez ²,³ , Luis Eduardo Pino ⁵
Resumen
El presente manuscrito revisa las aplicaciones actuales de la
inteligencia artificial (IA) en genómica
funcional. La reciente explosión de la IA sigue a los notables logros
que ha hecho posible el “aprendizaje profundo”, junto con una explosión
de “grandes conjuntos de datos” que pueden satisfacer su
necesidad. Esto ha sido posible gracias a los enormes avances en el
campo de las tecnologías de alto
rendimiento, aplicadas para determinar cómo los componentes
individuales de un sistema biológico
trabajan juntos para lograr diferentes procesos. Las disciplinas que
contribuyen a este volumen de datos se conocen colectivamente como
genómica funcional. Consisten en estudios de: i) la información
contenida en el ADN (genómica); ii) las modificaciones que el ADN puede
sufrir de forma reversible
(epigenómica); iii) las transcripciones de ARN originadas por un genoma
(transcriptómica); iv) el
conjunto de modificaciones químicas que decoran diferentes tipos de
transcripciones del ARN (epitranscriptómica); v) los productos de las
transcripciones que codifican proteínas (proteómica); y vi)
las pequeñas moléculas producidas a partir del metabolismo celular
(metabolómica) presentes en un
organismo o sistema en un momento dado, en condiciones fisiológicas o
patológicas. Palabras clave: Inteligencia artificial (IA); genómica; terapia; diagnóstico; medicina de precisión.
..............
² Fundación para la Investigación Clínica y Molecular Aplicada del Cáncer – FICMAC, Bogotá, Colombia.
³ Grupo de Investigación en Oncología Molecular y Sistemas Biológicos (Fox-G), Universidad El Bosque, Bogotá, Colombia.
⁴ Grupo Medicina Interna, Instituto Nacional de Cancerología – INC, Bogotá, Colombia.
⁵ Grupo Oncología Clínica, Instituto de Cáncer Carlos Ardila Lülle, Fundación Santa Fe de Bogotá, Bogotá, Colombia.
WALKING ON THE SHOULDERS OF GIANTS: THE INTERSECTION BETWEEN GENOMICS AND A
Abstract
The present manuscript reviews the current applications of artificial intelligence (AI) in functional genomics. The recent explosion of AI follows the remarkable achievements made possible by “deep learning”, along with a burst of “big data” that can meet its hunger. This has been made possible by huge advancements in the field of high throughput technologies, applied to determine how the individual components of a biological system work together to accomplish different processes. The disciplines contributing to this bulk of data are collectively known as functional genomics. They consist in studies of: i) the information contained in the DNA (genomics); ii) the modifications that DNA can reversibly undergo (epigenomics); iii) the RNA transcripts originated by a genome (transcriptomics); iv) the ensemble of chemical modifications decorating different types of RNA transcripts (epitranscriptomics); v) the products of protein-coding transcripts (proteomics); and vi) the small molecules produced from cell metabolism (metabolomics) present in an organism or system at a given time, in physiological or pathological conditions.Keywords: Artificial intelligence (AI); Genomics; Personalized treatment; Therapy; Diagnostic; Precision medicine.
Introducción
Pocas frases han resultado tan acertadas como el motivo primario de esta entrada “Caminamos a hombros de gigantes”. Este decálogo impreso en la revolución científica del siglo XVII y expuesto ampliamente en la Philosophiæ naturalis principia mathematica recogió de manera sencilla un apotegma de máximo calado. El sentido de la cita de Newton deja entrever que la dinámica de sus logros se apoyó, no sólo en sus propias virtudes, sino también en el conocimiento y saberes construidos por otros previamente. El origen de esta sentencia viene de una carta que escribió Isaac Newton a Robert Hooke el 15 de febrero de 1676, y proviene de un elocuente parafraseo a Bernard de Chartres, filósofo del siglo XII que había escrito “Somos como los enanos aupados a hombros de gigantes, de manera que podemos ver más cosas y más lejanas que ellos, no por la agudeza de nuestra vista o por nuestra elevada estatura, sino porque estamos alzados sobre ellos y nos elevamos sobre su gigantesca altura” (Según Jean de Salisbury). Simplificando la metáfora en la visión pragmática de John McCarthy, Marvin Minsky, Nat Rochester y Claude Shannon, la inteligencia artificial (IA) es la simulación de la razón en un agente no vivo. Posteriormente, y en el contexto diagnóstico se definió la IA como cualquier sistema o plataforma que permita interpretar adecuadamente diversos datos relacionados con salud, especialmente en su forma nativa. Globalmente, la mayoría de las tareas de interpretación de la IA se pueden agrupar por clases de problemas incluyendo la digitalización de imágenes, el análisis de series de tiempo, reconocimiento de voz, y el procesamiento del lenguaje natural, entre otros. Algunas de estas áreas tienen asociaciones diagnósticas que no parecen tan obvias, como la identificación de elementos reguladores del genoma a través de lectores visuales que permiten identificar lecturas anormales recurrentes en las secuencias de ADN, de forma análoga a la usada para la identificación de patrones de píxeles en imágenes por convolución (1,2).La IA se inspira en algoritmos in vivo, amplificando su capacidad de forma exponencial. Sin embargo, las aplicaciones de la IA en la genómica clínica están dirigidas a realizar tareas que resultan no funcionales para el utilitario humano debido a la propensión al error de los enfoques estadísticos estándares. Muchas de las herramientas de la IA se han adaptado para abordar múltiples pasos involucrados en el análisis genómico, incluido el llamado de variantes y su clasificación, el análisis de la correspondencia entre el fenotipo y genotipo, y eventualmente, predecir la modificación dinámica del fenotipo a partir de un genotipo base (3).
La interpretación clínica del genoma es sensible a la identificación de variantes individuales entre millones que constituyen el ecosistema de una célula entre millones de ellas, evento que requiere una precisión extrema. Las herramientas convencionales son propensas al equívoco sistemático asociado a la sutileza propia de la preparación de las muestras y librerías, a la tecnología utilizada para la secuenciación, al contexto de la secuencia, y por la influencia, muchas veces impredecible de la evolución biológica (mosIAcismo somático o cambios epigenéticos) (4). Los algoritmos generados por la IA pueden aprender los sesgos del análisis del genoma a partir de una fuente de variantes de referencia para producir estrategias adaptativas para el llamado de variantes. DeepVariant, una plataforma para el llamado de variantes basado en redes neuronales convolucionales demostró recientemente un mayor rendimiento para la identificación de variantes a partir de dependencias complejas en la secuenciación (5). Además, resultados recientes sugieren que el aprendizaje profundo (del inglés, deep learning) está listo para revolucionar la identificación de variantes para las tecnologías de secuenciación basadas en nanoporos (6). Hace poco, Luo y colaboradores demostraron la utilidad de la red neuronal convolucional ClIArvoyante, un modelo capaz de reducir el margen de error del análisis de una secuencia nucleotídica bajo el 5%. Este sistema disponible en código abierto obtuvo puntajes superiores al 90% para la predicción de variantes (incluyendo SNP o indels) obtenidas a partir de las plataformas Illimina, PacBio y Oxford Nanopore. El modelo de IA fue reproducible en muestras independientes y logró encontrar variantes en menos de 2 horas en un servidor estándar (7).
Clasificación de variantes
Se han desarrollado diversos métodos para la clasificación de variantes no sinónimas (8). Algunos, se han integrado en meta-predictores basados en aprendizaje profundo (modelos que procesan y fusionan las predicciones producidas por varios otros predictores) que superan tanto a sus componentes individuales como a la combinación de estos cuando se integran mediante una regresión. Por ejemplo, el enfoque combinado de agotamiento dependiente de anotaciones (CADD, por su denominación en inglés combined annotationdependent depletion approach) integra una variedad de características en un algoritmo de aprendizaje automático enfocado en predecir la patogenicidad de las variantes genéticas. Una extensión de CADD basada en aprendizaje profundo, denominada DANN (del ingles, Domain-Adversarial Training of Neural Networks), demostró un rendimiento mejorado utilizando el mismo conjunto de características de entrada que CADD, pero combinadas en una red neuronal profunda (9). No obstante, por el momento la precisión en la clasificación de las variantes no ha resultado suficiente para facilitar la interpretación de los informes clínicos. Otros métodos basados en IA hacen predicciones a partir de datos de sencuencias proteicas o de ADN con un mínimo de elaboración manual. El enfoque de Primate IA que utilizó una red neuronal convolucional usó información de especies cruzadas para facilitar el análisis. La red pudo aprender dominios de diferentes proteínas relevantes, aminoácidos conservados y mutaciones patogénicas con representatividad en múltiples enfermedades. PrimateIA integró su entrenamiento a partir de 120.000 muestras humanas y superó sustancialmente el rendimiento de otras herramientas de uso regular para la predicción de variantes patogénicas incluidas en Clinvar (10).Clasificación de variantes no codificantes
La identificación computacional y la predicción de la variación patogénica de regiones no codificantes sigue siendo un desafío abierto para la genómica humana. Hallazgos recientes sugieren que los algoritmos de IA mejorarán sustancialmente nuestra capacidad para comprender la variación genética de las regiones no codificantes. Los defectos en el empalme génico son responsables del 10% de la variación genética patogénica, suelen ser raros y difíciles de identificar debido a la complejidad de los potenciadores de empalme (splicing enhancers) intrónicos y exónicos. SpliceIA, es una red neuronal profunda de 32 capas capaz de predecir empalmes canónicos y no canónicos directamente a partir de los datos de una secuencia de unión exón-intrón (11) (Figura 1). Sorprendentemente, SpliceIA pudo usar información de una secuencia de largo alcance (long-range sequence) aumentar la precisión para predecir desde un 57%, utilizando un tamaño de ventana corto (80 nucleótidos) típico para muchas herramientas de predicción de empalme, hasta el 95% cuando se usó el algoritmo de IA. De igual forma, el modelo de IA fue capaz de identificar variantes candidatas de empalme críptico (ocultas) subyacentes a los trastornos del neurodesarrollo. Otro enfoque basado en aprendizaje profundo (DeepSEA) mejoró sustancialmente la capacidad para predecir la presencia de sitios hipersensibles a la ADNasa, diversas vías de transcripción y los cambios estructurales en las histonas (12). Como ejemplo, varias extensiones del modelo DeepSEA usadas en secuencias genómicas de familias con trastornos del espectro autista han revelado mutaciones de novo en segmentos no codificantes (13). En paralelo, extensiones del algoritmo ExPecto revelaron una mejoría en la capacidad pronóstica de diferentes perfiles de expresión génica extraídos a partir de secuencias de ADN germinal y somático (14).Mapeo fenotipo-genotipo
El genoma humano contiene numerosas variantes genéticas patogénicas o potencialmente patogénicas (15), independientemente del estado de salud del individuo estudiado (16). Los algoritmos de IA han permitido mejorar el mapeo fenotipo-genotipo, especialmente a través de la extracción de información derivada del diagnóstico clínico, la integración de imágenes y el uso de datos derivados de registros electrónicos de la historia clínica. Considerando la utilidad de las imágenes dentro del procesamiento del diagnóstico genético, el desarrollo de la estructura facial constituye un ejemplo perfecto. La ontología del fenotipo humano enumera 1.007 términos para las anomalías faciales; estas alteraciones están asociadas con 4.526 enfermedades y 2.142 genes. Un experto en dismorfología a menudo identificará estas anomalías de manera individual y las sintetizará en un diagnóstico puntual o la conjunción de estos. Es así como el diagnóstico clínico puede enfocar la secuenciación de genes específicos basado en el fenotipo dominante. A menudo, el diagnóstico clínico y los hallazgos moleculares se superponen, pero no coinciden con precisión debido a la similitud fenotípica de múltiples alteraciones nosológicas bien caracterizadas. DeepGestalt, es un algoritmo de análisis de imágenes faciales basado en redes neuronales convolucionales que supera la valoración clínica, siendo lo suficientemente preciso para distinguir entre diagnósticos moleculares asignados a un mismo diagnóstico (por decir algo, diversas formas moleculares del síndrome de Noonan) (17) (Figura 2). Al integrar DeepGestalt con PEDIA (sistema de interpretación del genoma que integrado), el modelo fue capaz de utilizar características fenotípicas extraídas de fotografías faciales para priorizar con precisión variantes patogénicas candidatas para 105 trastornos monogénicos diferentes en una población de 679 casos analizados (18). Estos hallazgos han permitido conjeturar acerca de la utilidad del DeepGestalt en la realización de escaneo facial dinámico facilitando la identificación de numerosos síndromes genéticos (19), entre otros, el síndrome de Cornelia de Lange, diagnosticado por IA con una exactitud del 98,6% (75-85% para la evaluación clínica), o el síndrome de Angelman determinado con una precisión del 92% (72% para el diagnóstico clínico) (17).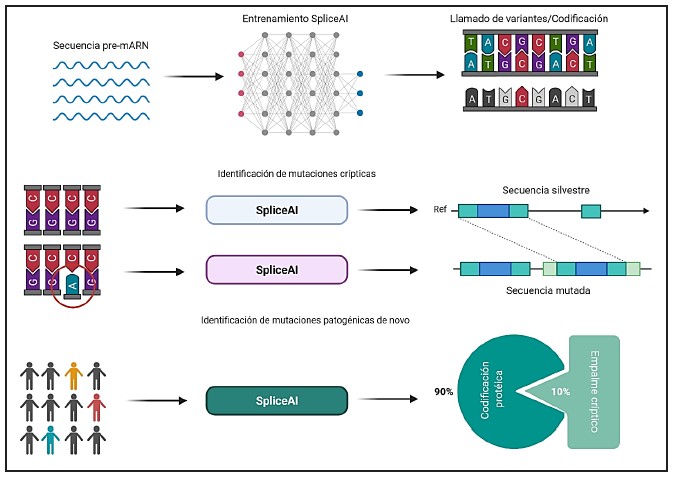
Figura 1. SpliceIA, una red neuronal profunda que modela con precisión el empalme de ARNm a partir de una secuencia genómica y predice la presencia de mutaciones de cripticas no codificantes en pacientes con enfermedades genéticas raras.
A pesar de la notable integración entre la genómica y la IA, aún persisten los sesgos propios de la selección en el proceso de aprendizaje. Por ejemplo, el DeepGestalt mostró precisión marginal para la identificación del síndrome de Down en individuos de ascendencia africana versus caucásicos europeos (36,8% frente a 80%, respectivamente) (18). El reentrenamiento del modelo con ejemplos del síndrome de Down en sujetos de raza negra mejoró el perfil diagnóstico con una precisión cercana al 95%, dejando claro que el rendimiento inicial es propenso a la desigualdad interpoblacional favorecida por la subrepresentación muestral en el grupo de entrenamiento (19).
Los síndromes genéticos que se identifican a través del análisis facial se pueden confirmar por el análisis genómico; sin embargo, en el caso del cáncer, el fenotipo tumoral o las imágenes diagnósticas son incapaces de priorizar y predecir el patrón de las alteraciones somáticas del ADN. La IA ha permitidos disminuir la brecha entre el análisis fenotípico derivado de las imágenes y el origen genómico de las neoplasias sólidas. Un modelo de red neuronal convolucional integrado con datos de supervivencia pudo reconocer las características histológicas de los tumores cerebrales gliales, prediciendo las alteraciones canónicas y la respuesta al tratamiento (20). De forma general, algunos sistemas de visión computarizada basados en IA pueden predecir las aberraciones genómicas presentes en individuos con fenotipos complejos integrados en imágenes relevantes (20).
De la historia clínica electrónica al diagnóstico genético
El fenotipo de algunas enfermedades puede ser complejo y presentar un evolutivo multidimensional. Estas diferencias suelen ser capturadas por las imágenes diagnósticas, diferentes análisis bioquímicos, y el uso de biomarcadores de precisión, ensayos que se pueden ser solicitados en diferentes momentos de la enfermedad y por diversos profesionales en el curso de la identificación de un diagnóstico diferencial (21). Estos resultados se documentan en la historia clínica electrónica donde se sintetizan los hallazgos para facilitar la toma de decisiones y comunicar las decisiones clínicas. Los sistemas de IA facilitan el reconocimiento de patrones en estos registros. Un estudio reciente que involucró más de 500.000 pacientes utilizó un modelo para procesamiento natural del lenguaje (Natural language processing) basado en IA para extraer características clínicamente relevantes de la información consignada (22). Después de entrenar al modelo estadístico jerárquico y escalonado sobre la base de divisiones anatómicas se generó un sistema diagnóstico adaptado capaz de diferenciar 55 diagnósticos pediátricos comunes con un 92% de precisión. En paralelo, el uso del modelo vinculado a información genómica permitió estratificar enfermedades raras en los niños y clasificar coincidencias con variantes patogénicas extraídas del análisis del genoma de los pacientes (23). En 101 niños con 105 enfermedades genéticas, la valoración retrospectiva de los diagnósticos genómicos por vía automatizada coincidió con la interpretación humana experta con un 99% de precisión (24).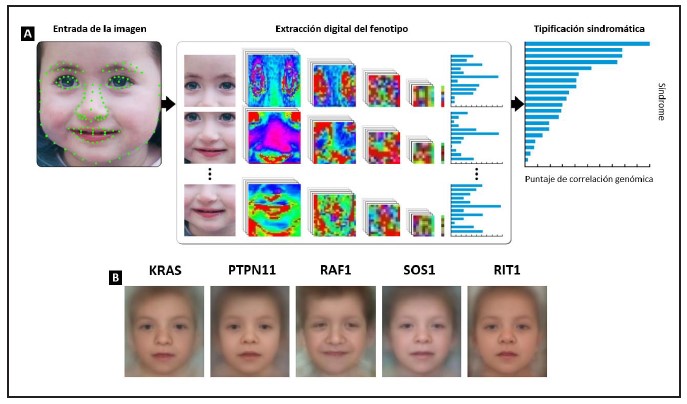
Figura 2A. DeepGestalt, flujo de alto nivel. La imagen de entrada se procesa primero para lograr una detección del perfil facial, para la detección de puntos de referencia y su alineación. Después del preprocesamiento, la imagen de entrada se recorta en regiones. Luego, cada región alimenta una red neuronal convolucional para obtener un vector softmax que indica su correspondencia con cada síndrome en el modelo. Los vectores de salida de cada estrato de la red neuronal convolucional se agregan y clasifican para obtener la lista que se correlaciona con el patrón genómico. El histograma del lado derecho representa los síndromes de salida del DeepGestalt, ordenados por la puntuación de similitud. B. Fotografías compuestas de pacientes con síndrome de Noonan con diferentes genotipos y diferencias faciales sutiles caracterizados a través del DeepGestalt. Foto publicada con el consentimiento de los padres y reproducida con autorización de Gurovich Y, Hanani Y, Bar O, et al. Identifying facial phenotypes of genetic disorders using deep learning. Nat Med. 2019 Jan;25(1):60-64 (17).
Desafíos en la interpretación de la genómica tumoral
La secuenciación de próxima generación (NGS, Next Generation Sequencing) ha revolucionado la investigación biomédica permitiendo la generación de estudios multicapa que integran datos genómicos en diversas dimensiones, incluyendo el ADN-seq y la ARN-seq, así como información multiómica que incluye al proteoma, epigenoma, metaboloma, microbioma, etc (25). Esta fusión proporciona una visión más completa de los procesos y sistemas biológicos, lo que conduce a una mejor comprensión de la enfermedad, especialmente en comparación con el análisis de una sola capa. Sin embargo, existen varios desafíos para la traducción de datos multiómicos en biomarcadores clínicamente procesables. Primero, combinar perfiles de datos en varios niveles daría como resultado una alta dimensionalidad con un gran número de covariables. La escasez de datos de alta dimensionalidad asociada con la alta heterogeneidad entre los diversos tipos de datos impone una dificultad significativa en los análisis integrativos. Se han desarrollado diferentes técnicas para reducir estas dimensiones, incluyendo el análisis de co-inercia múltiple y el análisis de factores múltiples, diseños útiles para facilitar los análisis en la transcripción corriente abajo (down stream signaling). Varios marcos se han utilizado para la integración, incluyendo los enfoques basados en redes que utilizan algoritmos gráficos para capturar interacciones potenciales entre las redes moleculares, y modelos bayesianos de varios niveles que imponen suposiciones realistas para la estimación de parámetros a través de una estructura de entrada y salida (26,27). En segundo lugar, la integración de datos multiómicos requiere la mejor progresiva de los estándares para la generación de resultados, facilitando la interpretación y reduciendo el sesgo.Por otra parte, los procedimientos para la adquisición y preparación de las muestras deben estar correctamente regulados para cada una de las plataformas de secuenciación y entre ellas. Por ejemplo, para la información derivada del análisis por NGS se necesita material de referencia (CLSI QMS01-A 2018; CLSI MM01A3E 2018; NIST 2018) cuyas propiedades sean suficientemente homogéneas y estén bien establecidas para la calibración del sistema de secuenciación. Por último, pero no menos importante, se necesitan estudios bien diseñados que permitan hacer inferencia causal para filtrar los biomarcadores que tienen fuertes efectos predictivos (28).
La diversidad de la evidencia puede contribuir con la inferencia patogénica de las variantes, incluidos datos genéticos, informáticos y experimentales. A nivel genético, las variantes patogénicas pueden enriquecerse significativamente a partir del análisis de casos y controles y/o ante la evidencia de una variante germinal que afecta el estado de la enfermedad dentro de una familia afecta. En el nivel informático, las variantes patogénicas se pueden encontrar en el lugar que se predice que causará una alteración funcional (región de unión a proteínas). Y a nivel experimental, las variantes patogénicas pueden alterar significativamente los niveles, el empalme o la función bioquímica normal del producto de los genes afectados. Esto puede mostrarse en células de pacientes o bien puede ser validado con modelos in vitro o in vivo (29,30).
El avance de las tecnologías relacionadas con machine learning está destinado a afectar la interpretación de los datos provenientes de la secuenciación genómica, que tradicionalmente se basó en la curación manual. Estos esfuerzos de purificación se basan en la estructura de proteínas, estudios funcionales y, más recientemente, en modelos “in silico” que predicen el impacto funcional de la alteración genética usando plataformas como SIFT, PANTHER-PSEP, PolyPhen2 y otros (30). En adición, las bases de datos genómicas como ClinVar, COSMIC y OncoKB han proliferado como medio para compilar de manera concisa una colección de las variantes genéticas (Figura 3). En general, proporcionan la evidencia que respalda la clasificación de una variante como patogénica, benigna o de significado desconocido (VUS).
Dos de las limitaciones clave de la curación e interpretación manual de los resultados derivados de datos genómicos crudos son la escalabilidad y la reproducibilidad. Estos desafíos continúan creciendo a medida que se dispone de más información. La cantidad de expertos en clasificación de variantes y la cantidad de tiempo que pueden dedicar diariamente a esta tarea es limitada. Para abordar estas limitaciones, varias organizaciones están trabajando en crear y estandarizar protocolos para la clasificación de variantes, incluyendo el American College of Medical Genetics and Genomics y la Association for Molecular Pathology (ACMG-AMP), quienes ya publicaron una serie de directrices para la interpretación de variantes genéticas de la línea germinal y somática para genes causantes de trastornos hereditarios y del cáncer (31,32). Sin embargo, la capacidad de escalar la interpretación de variantes provenientes de los estudios de NGS, especialmente en cáncer, sigue siendo limitada, requiere validación y un estricto control de calidad (33). Recientemente se presentó la plataforma OncoTree que incluye 886 tipos de tumores originados en 32 complejos tisulares; esta plataforma fue adoptada como sistema de clasificación para el proyecto Genomics Evidence Neoplasia Information Exchange (GENIE) de la Asociación Estadounidense para la Investigación del Cáncer (AACR), un gran consorcio de intercambio de datos genómicos y clínicos, para amplificar y unificar el esfuerzo de OncoKB y cBioPortal for Cancer Genomics (34).
Cómo la integración de la NGS y la IA están cambiando el panorama de la genómica tumoral
Actualmente, la NGS se aplica ampliamente como método valioso para obtener un perfilamiento genómico exhaustivo. Gracias a esta tecnología se ha logrado secuenciar simultáneamente millones de fragmentos de ADN en una sola muestra para detectar una amplia gama de aberraciones propias del cáncer. Los paneles de cáncer están diseñados específicamente para detectar mutaciones somáticas y germinales clínicamente relevantes. De igual forma, la caracterización molecular usando NGS por biopsia líquida facilita el diagnóstico temprano, la evaluación de la heterogeneidad tumoral y de la enfermedad mínima residual siguiendo un principio no invasivo. Gracias a estos modelos de tipificación genómica se abrieron proyectos como el Cancer Genome Atlas (por su sigla en inglés, TCGA) que ha permitido el descubrimiento de nuevos mecanismos oncogénicos y la estratificación de pacientes y enfermedades (35). La información utilizada para dilucidar los mecanismos primarios en la evolución del cáncer ha permitido elucidar el metabolismo oxidativo de las células tumorales (36), validar la utilidad de biomarcadores predictivos como la metilación del promotor de la MGMT en tumores de estirpe glial (37), proyectar el efecto terapéutico de la inmunoterapia en cáncer gástrico (38), confirmar la utilidad in silico de mutaciones particulares en cáncer de pulmón (38), y considerar a la transición epitelio mesenquimal como parte de la resistencia en cáncer de seno (39), entre muchos otros.
Figura 3. Repertorio de plataformas que integran IA para el llamado y análisis de variantes génicas.
Uno de los mayores retos de la genómica tumoral está asociado con el llamado, depuración e interpretación de las variantes. Frecuentemente, los usuarios necesitan ajustar los parámetros de forma heurística y aplicar filtros personalizados para eliminar los falsos positivos antes de lograr una precisión aceptable. Este es un esfuerzo que requiere tiempo y experiencia para ajustar las puntuaciones de calidad y los atributos dentro de los contextos de secuenciación. Diferentes grupos están aprovechando algoritmos de aprendizaje automático y el entrenamiento en las características de calidad subyacentes para mejorar el rendimiento del llamado de variantes, especialmente en escenarios subóptimos (40-43). Estos, han permitido establecer el valor de la ploidia como factor que contribuye con la complejidad de la enfermedad. De igual forma, contribuyeron para establecer el valor de las variantes subclonales (presentes solo en unas pocas células), difíciles de detectar porque su representación en la librería de secuenciación suele ser baja. Este hallazgo resulta en una mayor variabilidad entre los métodos de análisis, los umbrales y las puntuaciones de calidad que pueden no ser lo suficientemente flexibles para detectar la evolución subclonal de la enfermedad (41). En lugar de configurar reglas estáticas, los métodos de IA permiten ajustar los umbrales de forma dinámica en función de los patrones de expresión génica. Las variantes con frecuencias alélicas muy bajas aún se pueden informar si la profundidad de secuenciación y otras métricas de calidad superan los umbrales. Por ejemplo, un modelo de red neuronal convolucional cuyos algoritmos se utilizan a menudo en el reconocimiento de imágenes logró una puntuación F1 de 0,96 y pudo alcanzar variantes con una frecuencia de alelos tan baja como 0,0001 (la puntuación F1 es una medida que tiene en cuenta tanto la precisión como la memoria) (44). En otro caso, un enfoque basado en machine learning aplicado a los datos de NGS mostró una precisión mejorada (medida por la puntuación F1) en la identificación de mutaciones tumorales en comparación con otros programas existentes como MuTect1, MuTect2, SomaticSniper, Strelka, VarDict y VarScan2. Si bien sus valores de recuperación fueron similares, la plataforma de IA mostró mayor precisión (45). Se han descrito éxitos similares para el análisis de variación del número de copias (CNV) (46,47).
Además de los paradigmas para la detección de variantes estándar, DeepVariant de Google transformó un problema de convencional en otro para el reconocimiento de imágenes al convertir un archivo BAM en imágenes similares a las instantáneas del navegador del genoma, donde el llamado de variantes se hace utilizando el marco para un flujo de tensor de inicio (del ingles, Inception Tensor Flow) que se desarrolló originalmente para la clasificación visual computarizada (48). Otro estudio reciente aplicó con éxito el machine learning para la secuenciación de datos de múltiples regiones de un tumor, permitiendo identificar y aprender patrones de crecimiento como predictores precisos de la progresión del tumor (49). Adicionalmente, se están entrenando otros modelos de IA para caracterización de estructuras secundarias incluyendo fosforilación proteica en respuesta a la administración de medicamentos (contemplando la dosis biológica efectiva) (50). Finalmente, el proceso de depuración depende de la homologación de decisiones de la IA a partir de nociones clínicas con enorme variabilidad intra e interindividual (Figura 4). Para validar el papel de múltiples modelos de aprendizaje de máquina (del inglés, machine learning), el hospital Mash General diseñó un estudio que incluyó ~500 características clínicas y cerca de 20.000 variantes somáticas con potencial en la toma de decisiones. La comparación de la estructura de IA contra la mesa multidisciplinaria de discusión genómica (del inglés, Genomic Tumor Board) demostró que el uso de una escala basada en regresión logística tuvo una tasa de falsos negativos y positivos del 1 y 2%, respectivamente, hallazgo que resultó comparable a las decisiones humanas (51).
Por otra parte, el volumen de literatura médica relacionado con genómica tumoral resulta inmanejable (~165.000/año). Esta dimensión podría llegar a ser manejable usando herramientas que contemplen el procesamiento natural del lenguaje para reducir el tiempo y esfuerzo necesarios para la recuperación de la información que permita la generación de nuevas hipótesis basadas en la mejor evidencia (Figura 5). La minería de datos también ha permitido el reconocimiento de entidades a través de proceso digitales de nominación 8Bio-NER) facilitando la extracción de referencias en medicina de precisión.
Desafortunadamente, no existe un estándar universal para denominar las variantes genéticas y existen múltiples formas de presentar el mismo evento en la literatura y en las bases de datos genómicas. Para consolidar el conocimiento sobre variantes patogénicas a partir de la literatura e integrarlas con los datos curados en recursos existentes como ClinVar y COSMIC, resulta esencial el uso correcto de la nomenclatura HGVS así como la introducción del número de identificación del SNP (variante de un solo nucleótido) de referencia (RSID) (52). Recientemente, se han aplicado varios métodos de aprendizaje profundo al reconocimiento de entidades con nombre biomédico y sus respectivas alteraciones genéticas con una ganancia significativa en el rendimiento para integrar mejor las características multidimensionales y, al mismo tiempo, minimizar los requerimientos manuales (53).
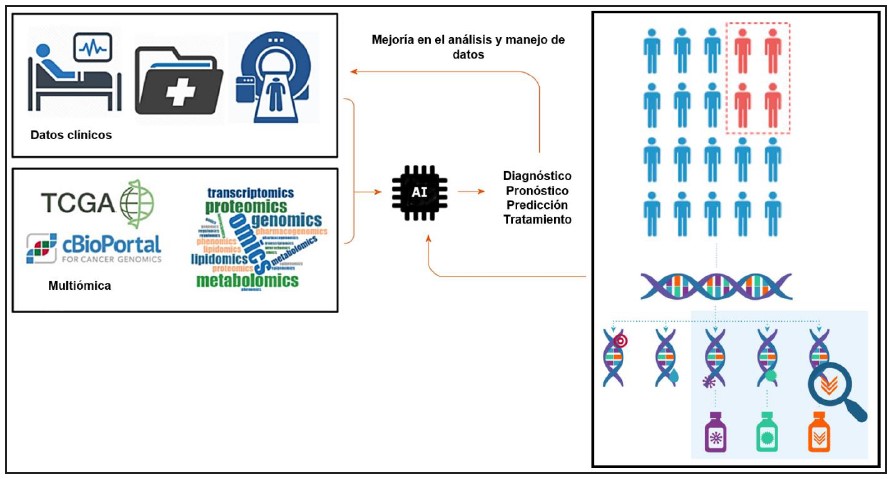
Figura 4. Interacción dinámica desde la clínica (información relacional de la sintomatología, imágenes y características del paciente integradas en la historia clínica electrónica) hacia la genómica y bioinformática a través de algoritmos de análisis multicapa que permiten la subselección de intervenciones terapéuticas basadas en medicina de precisión.
Retos para la implementación y uso de la IA en el ámbito de la genómica
La evaluación de la precisión de la IA en relación con la genómica es fundamental para titular el funcionamiento de los sistemas solventando el precepto de la “caja negra”. En la genómica tiene especial importancia la clasificación de variantes y su relevancia clínica, la validación de la literatura y la clasificación de vectores que permiten el diseño de biomarcadores. A pesar de la abundancia de información clínica y cruda de datos genómicos, la protección individual de estos documentos por las pautas HIPPA (Health Insurance Portability and Accountability Act) y GDPR (Reglamento General de Protección de Datos) limita el acceso a su estudio y uso para la capacitación y evaluación de los sistemas de IA aplicables al diseño de planes personalizados de tratamiento. En adición, la reproducibilidad de los resultados experimentales incluidos en los estudios de IA sigue siendo un problema para la implementación en la práctica clínica regular. Debido a que los algoritmos de aprendizaje suelen tener múltiples componentes ajustables, el rendimiento suele verse afectado por la sensibilidad de la escala y calidad de los datos de entrenamiento, la configuración empírica de los parámetros, y los procesos de inicialización y optimización. Muchas publicaciones no revelan los supuestos simplificadores o los detalles de implementación y, por lo tanto, dificultan la reproducción de los resultados. Finalmente, la mayoría de los estudios no comparte el código fuente.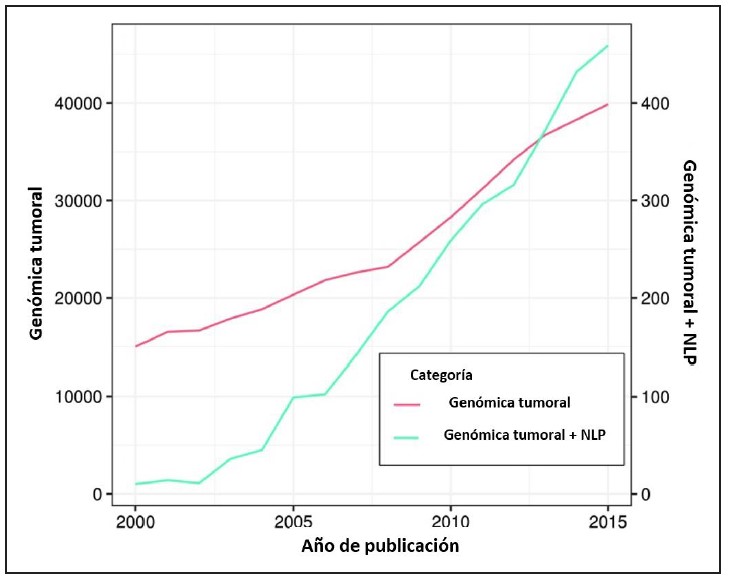
Figura 5. Número de publicaciones frente al año de indexación. En esta figura se representan dos ejes “y”, uno exhibe el número de artículos relacionados con genómica tumoral, y el otro, el número de manuscritos asociados con genómica más NLP. El eje “x” representa el año de publicación. Tomado y modificado con autorización de Xu J, Yang P, Xue S, et al. Translating cancer genomics into precision medicine with artificial intelligence: applications, challenges and future perspectives. Hum Genet. 2019 Feb;138(2):109-124. doi: 10.1007/s00439-019-01970-5.
Conclusión
Si bien la salud digital se ha vuelto esencial para brindar las mejores prácticas en el cuidado sanitario, plantea desafíos sin precedentes para los pacientes, investigadores y para la comunidad biomédica, en especial, cuando confluye con la complejidad de la medicina de precisión y los análisis multiómicos. Por el momento, la intersección entre la IA y la genómica semeja a gigantes entre los gigantes, recordando la respuesta que algún día diera Isaac Asimov a la pregunta sobre el científico más grande de la historia. Después de quedarse unos segundos en silencio, replicó entre dientes “La historia probablemente aún no lo ha visto, sin embargo, tengo dudas sobre a quién colocar en segundo lugar”. Entonces, Asimov consideraba que para este lugar ya había una dura liza entre Albert Einstein, Ernest Rutherford, Niels Borh, Louis Pasteur, Charles Darwin, Galileo Galilei, Arquímedes y algunos otros. Lo que sí tenía claro, era que al menos hasta donde su visión alcanzó, el mayor talento había sido de Isaac Newton. La IA transformará la historiografía de la biología molecular aplicada, en especial, para patologías complejas como el cáncer, donde la fuente del análisis avanzado de datos ya lo ha hecho y lo seguirá haciendo. Nada ha sido más estimulante que tener la oportunidad de vivirlo, nada vale más que reconocer que “antes pensábamos que nuestro futuro estaba en las estrellas, ahora sabemos que está en nuestros genes” (James Watson), y la IA está al servicio de la curiosidad para leerlos.Referencias
1.
Torkamani A, Andersen KG, Steinhubl SR, Topol EJ. High-definition medicine. Cell. 2017;170:828–43.2. Leung MKK, Xiong HY, Lee LJ, Frey BJ. Deep learning of the tissue-regulated splicing code. Bioinformatics. 2014;30:i121–9.
3. Jaganathan K, Kyriazopoulou Panagiotopoulou S, McRae JF, Darbandi SF, Knowles D, Li YI, et al. Predicting splicing from primary sequence with deep learning. Cell. 2019;176:535–48.
4. Quang D, Xie X. DanQ: a hybrid convolutional and recurrent deep neural network for quantifying the function of DNA sequences. Nucleic Acids Res. 2016;44:e107.
5. Poplin R, Chang PC, Alexander D, Schwartz S, Colthurst T, Ku A, et al. A universal SNP and small-indel variant caller using deep neural networks. Nat Biotechnol. 2018;36:983–7.
6. Wick RR, Judd LM, Holt KE. Performance of neural network basecalling tools for Oxford nanopore sequencing. Genome Biol. 2019;20:129.
7. Luo R, Sedlazeck FJ, Lam TW, Schatz MC. A multi-task convolutional deep neural network for variant calling in single molecule sequencing. Nat Commun. 2019 Mar 1;10(1):998.
8. Tang H, Thomas PD. Tools for predicting the functional impact of nonsynonymous genetic variation. Genetics. 2016;203:635–47.
9. Quang D, Chen Y, Xie X. DANN: a deep learning approach for annotating the pathogenicity of genetic variants. Bioinformatics. 2015;31:761–3.
10. Landrum MJ, Lee JM, Benson M, Brown GR, Chao C, Chitipiralla S, et al. ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018;46:D1062–7.
11. FDA approves stroke-detecting AI software. Nat Biotechnol. 2018;36:290.
12. Zhou J, Troyanskaya OG. Predicting effects of noncoding variants with deep learning-based sequence model. Nat Methods. 2015;12:931–4.
13. Zhou J, Park CY, Theesfeld CL, Wong AK, Yuan Y, Scheckel C, et al. Whole-genome deep-learning analysis identifies contribution of noncoding mutations to autism risk. Nat Genet. 2019;51:973–80.
14. Zhou J, Theesfeld CL, Yao K, Chen KM, Wong AK, Troyanskaya OG. Deep learning sequence-based ab initio prediction of variant effects on expression and disease risk. Nat Genet. 2018;50:1171–9.
15. Telenti A, Pierce LCT, Biggs WH, Di Iulio J, Wong EHM, Fabani MM, et al. Deep sequencing of 10,000 human genomes. Proc Natl Acad Sci U S A. 2016;113:11901–6.
16. Erikson GA, Bodian DL, Rueda M, Molparia B, Scott ER, Scott-Van Zeeland AA, et al. Whole-genome sequencing of a healthy aging cohort. Cell. 2016;165:1002–11.
17. Gurovich Y, Hanani Y, Bar O, Nadav G, Fleischer N, Gelbman D, et al. Identifying facial phenotypes of genetic disorders using deep learning. Nat Med. 2019;25:60–4.
18. Lumaka A, Cosemans N, Lulebo Mampasi A, Mubungu G, Mvuama N, Lubala T, et al. Facial dysmorphism is influenced by ethnic background of the patient and of the evaluator. Clin Genet. 2017;92:166–71.
19. Martin AR, Kanai M, Kamatani Y, Okada Y, Neale BM, Daly MJ. Clinical use of current polygenic risk scores may exacerbate health disparities. Nat Genet. 2019;51:584–91.
20. Hsieh T-C, Mensah MA, Pantel JT, Aguilar D, Bar O, Bayat A, et al. PEDIA: prioritization of exome data by image analysis. Genet Med. 2019.
21. Dolgin E. AI face-scanning app spots signs of rare genetic disorders. Nature. 2019.
22. Mobadersany P, Yousefi S, Amgad M, Gutman DA, Barnholtz-Sloan JS, Velázquez Vega JE, et al. Predicting cancer outcomes from histology and genomics using convolutional networks. Proc Natl Acad Sci U S A. 2018;115:E2970–9.
23. Clark MM, Hildreth A, Batalov S, Ding Y, Chowdhury S, Watkins K, et al. Diagnosis of genetic diseases in seriously ill children by rapid whole-genome sequencing and automated phenotyping and interpretation. Sci Transl Med. 2019;11:eaat6177.
24. Lello L, Avery SG, Tellier L, Vazquez AI. de los Campos G, Hsu SDH. Accurate genomic prediction of human height. Genetics. 2018;210:477–97.
25. Wang B, Mezlini AM, Demir F, et al. Similarity network fusion for aggregating data types on a genomic scale. Nat Methods. 2014 Mar; 11(3):333-7.
26. Meng C, Zeleznik OA, Thallinger GG, et al. Dimension reduction techniques for the integrative analysis of multiomics data. Brief Bioinform. 2016 Jul; 17(4):628-41.
27. Bersanelli M, Mosca E, Remondini D, et al. Methods for the integration of multi-omics data: mathematical aspects. BMC Bioinformatics. 2016 Jan 20; 17 Suppl 2:15.
28. Ibrahim R, Pasic M, Yousef GM. Omics for personalized medicine: defining the current we swim in. Expert Rev Mol Diagn. 2016 Jul;16(7):719-22.
29. MacArthur DG, Manolio TA, Dimmock DP, et al. Guidelines for investigating causality of sequence variants in human disease. Nature. 2014 Apr 24; 508(7497):469-76.
30. Tang H, Thomas PD. Tools for Predicting the Functional Impact of Nonsynonymous Genetic Variation. Genetics. 2016 Jun; 203(2):635-47.
31. Richards S, Aziz N, Bale S, et al; ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015 May;17(5):405-24.
32. Li MM, Datto M, Duncavage EJ, et al. Standards and Guidelines for the Interpretation and Reporting of Sequence Variants in Cancer: A Joint Consensus Recommendation of the Association for Molecular Pathology, American Society of Clinical Oncology, and College of American Pathologists. J Mol Diagn. 2017 Jan;19(1):4-23.
33. Lindeman NI, Cagle PT, Aisner DL, et al. Updated Molecular Testing Guideline for the Selection of Lung Cancer Patients for Treatment With Targeted Tyrosine Kinase Inhibitors: Guideline From the College of American Pathologists, the International Association for the Study of Lung Cancer, and the Association for Molecular Pathology. J Mol Diagn. 2018 Mar;20(2):129-159. doi: 10.1016/j.jmoldx.2017.11.004. Epub 2018 Jan 23.
34. Kundra R, Zhang H, Sheridan R, et al. OncoTree: A Cancer Classification System for Precision Oncology. JCO Clin Cancer Inform. 2021 Feb;5:221-230. doi: 10.1200/CCI.20.00108.
35. Weinstein JN, Collisson EA, Mills GB, et al; Cancer Genome Atlas Research Network. The Cancer Genome Atlas Pan-Cancer analysis project. Nat Genet. 2013 Oct; 45(10):1113-20.
36. Davis RJ, Gönen M, Margineantu DH, et al. Pan-cancer transcriptional signatures predictive of oncogenic mutations reveal that Fbw7 regulates cancer cell oxidative metabolism. Proc Natl Acad Sci U S A. 2018 May 22;115(21):5462-5467.
37. Castro M, Pampana A, Alam A, et al. Combination chemotherapy versus temozolomide for patients with methylated MGMT (m-MGMT) glioblastoma: results of computational biological modeling to predict the
magnitude of treatment benefit. J Neurooncol. 2021 Jul;153(3):393-402. doi: 10.1007/s11060-021-03780-0.
38. Zhang Z, He T, Huang L, et al. Immune gene prognostic signature for disease free survival of gastric cancer: Translational research of an artificial intelligence survival predictive system. Comput Struct Biotechnol J. 2021 Apr 12;19:2329-2346. doi: 10.1016/j.csbj.2021.04.025.
39. Nosi V, Luca A, Milan M, et al. MET Exon 14 Skipping: A Case Study for the Detection of Genetic Variants in Cancer Driver Genes by Deep Learning. Int J Mol Sci. 2021 Apr 19;22(8):4217. doi: 10.3390/ijms22084217.
40. Chakraborty D, Ivan C, Amero P, et al. Explainable Artificial Intelligence Reveals Novel Insight into Tumor Microenvironment Conditions Linked with Better Prognosis in Patients with Breast Cancer. Cancers (Basel). 2021 Jul 9;13(14):3450. doi: 10.3390/cancers13143450.
41. Ding J, Bashashati A, Roth A, et al. Feature-based classifiers for somatic mutation detection in tumour-normal paired sequencing data. Bioinformatics. 2012 Jan 15; 28(2):167-75.
42. Hao Y, Xuei X, Li L, et al. RareVar: A Framework for Detecting Low-Frequency Single-Nucleotide Variants. J Comput Biol. 2017 Jul;24(7):637-646.
43. Spinella JF, Mehanna P, Vidal R, et al. SNooPer: a machine learning-based method for somatic variant identification from low-pass next-generation sequencing. MC Genomics. 2016 Nov 14;17(1):912.
44. ST, et al (2018) Deep learning mutation prediction enables early-stage lung cancer detection in liquid biopsy. IN: ICLR 2018 conference, Vancouver.
45. Wood DE, White JR, Georgiadis A, et al. A machine learning approach for somatic mutation discovery. Sci Transl Med. 2018 Sep 5;10(457):141.
46. Antaki D, Brandler WM, Sebat J. SV2: accurate structural variation genotyping and de novo mutation detection from whole genomes. Bioinformatics. 2018 May 15; 34(10):1774-1777.
47. Onsongo G, Baughn LB, Bower M, et al. CNV-RF Is a Random Forest-Based Copy Number Variation Detection Method Using Next-Generation Sequencing. J Mol Diagn. 2016 Nov; 18(6):872-881.
48. Going Deeper with Convolutions (2014) arXiv:1409.4842v1.
49. Caravagna G, Giarratano Y, Ramazzotti D, et al. Detecting repeated cancer evolution from multi-region tumor sequencing data. Nat Methods. 2018 Sep;15(9):707- 714.
50. Qi H, Zhang H, Zhao Y, et al. MVP predicts the pathogenicity of missense variants by deep learning. Nat Commun. 2021 Jan 21;12(1):510. doi: 10.1038/s41467-020- 20847-0.
51. Zomnir MG, Lipkin L, Pacula M, et al. Artificial Intelligence Approach for Variant Reporting. JCO Clin Cancer Inform. 2018;2:CCI.16.00079. doi: 10.1200/CCI.16.00079. Epub 2018 Mar 22.
52. Krallinger M, Vazquez M, Leitner F, et al. The ProteinProtein Interaction tasks of BioCreative III: classification/ ranking of articles and linking bio-ontology concepts to full text. BMC Bioinformatics. 2011 Oct 3;12 Suppl 8(Suppl 8):S3. doi: 10.1186/1471-2105-12-S8-S3.
53. Habibi M, Weber L, Neves M, et al. Deep learning with word embeddings improves biomedical named entity recognition. Bioinformatics. 2017 Jul 15;33(14):i37-i48..
Recibido: 12 de Noviembre, 2021
Aceptado: 22 de Noviembre, 2021
Correspondencia:
Andrés F. Cardona
acardona@fctic.org