APLICACIONES DE LA INTELIGENCIA ARTIFICIAL EN LA FARMACOLOGÍA BÁSICA Y CLÍNICA
..................
Christos Matsingos ¹,², Ana María Urdaneta ³, Juan Camilo Hernández ⁴, Ricardo A. Peña-Silva ¹,⁵
Resumen
El descubrimiento y desarrollo de fármacos es un proceso
complicado y arduo que implica un gran
esfuerzo interdisciplinar. A grandes rasgos, el proceso puede dividirse
en dos partes: la preclínica y la
clínica, que juntas pueden durar hasta 12 años y costar entre 2 y 3
billones de dólares. Debido a la compleja naturaleza de la creación de
nuevos medicamentos, que implica consideraciones bioquímicas y
fisicoquímicas, así como consideraciones de seguridad y eficacia
clínica, el proceso de descubrimiento
de fármacos se caracteriza por una alta tasa de fracasos. En la era de
la información, este proceso de
desarrollo suele estar asociado a la generación de grandes cantidades
de datos. La inteligencia artificial
ha permitido aprovechar estos datos para acelerar el proceso de
descubrimiento de fármacos y evitar
posibles escollos que puedan llevar al fracaso de la comercialización
de un medicamento. En esta revisión analizamos los nuevos avances en
inteligencia artificial y aprendizaje automático en diferentes
partes del proceso de descubrimiento de fármacos, desde la síntesis
química hasta la selección de candidatos para los ensayos clínicos. Se
muestra que la inteligencia artificial se ha aplicado en todas las
etapas del descubrimiento de fármacos y se ha utilizado en gran medida
para revolucionar los métodos
de investigación tradicionales. La inteligencia artificial no sólo se
ha utilizado para facilitar y acelerar
los procesos de descubrimiento, sino también para obtener conocimientos
y detectar patrones que no
se conocían antes. El uso de inteligencia artificial es indispensable
para el futuro del descubrimiento
de fármacos. Palabras clave: Farmacia; análisis de datos; eficacia; inteligencia artificial; medicamentos.
..............
² School of Physical and Chemical Sciences, Queen Mary University of London, Londres, Reino Unido.
³ MD. MSc. Miembro fundador de AIpocrates, Comité de analítica y calidad del dato.
⁴ QF. MSc. Universidad CES, Medellín, Colombia.
⁵ MD, PhD. Lown Scholars Program, Harvard T.H. Chan School of Public Health, Boston, Massachusetts, USA.
APPLICATIONS OF ARTIFICIAL INTELLIGENCE IN BASIC AND CLINICAL PHARMACOLOGY
Abstract
Drug discovery and development is a complicated and arduous process that involves a great interdisciplinary effort. The process can be broadly split into two parts: the preclinical and the clinical, that together can take up to 12 years and cost around 2-3 billion dollars to complete. Due to the complex nature of creating new medicines which involves biochemical and physicochemical considerations, as well as, considerations of clinical safety and efficacy, the drug discovery pipeline is characterised by high attrition rates. In the information age this process is often associated with the generation of large amounts of data. Artificial intelligence has allowed us to harness this data to speed up the process of drug discovery and avoid possible pitfalls that may lead to the failure of taking a drug to market. In this review we discuss new advances in artificial intelligence and machine learning in different parts of the drug discovery process, ranging from chemical synthesis to candidate selection for clinical trials. We show that artificial intelligence has been applied in every part of drug discovery and has been largely used to revolutionise traditional research methods. It has not just been used to facilitate and speed up discovery processes but also to gain insights and detect patterns that have not been known before. The use of artificial intelligence is indispensable for the future of drug discovery.Keywords: Pharmacy; Data Analysis; Efficacy; Artificial Intelligence
Introducción
Conceptos básicos de la inteligencia artificial
La digitalización de los datos ha tomado un papel importante en la sociedad actual y cada vez es más común ver cómo en los diferentes campos se han aplicado herramientas de la inteligencia artificial para procesar grandes volúmenes de datos y optimizar la toma de decisiones (1,2). La inteligencia artificial requiere la participación e integración de conocimientos de diferentes disciplinas como la matemática, estadística, informática, economía, entre otras. En la actualidad, las aplicaciones de la inteligencia artificial han pasado de la teoría al mundo real, y esto ha permitido notorios avances en los procesos de diversas áreas, incluyendo las ciencias de la salud y la farmacología (3-5).La farmacología es la ciencia que estudia los efectos e interacciones entre los fármacos y sistemas biológicos. Un fin de la farmacología es encontrar moléculas que modulan la actividad de un blanco biológico, para alterar el curso de una enfermedad (6). Para que un fármaco sea útil en humanos o en organismos (actividad clínica), es necesario que se identifiquen blancos biológicos relevantes en la fisiopatología de una enfermedad, que existan moléculas que puedan modular la actividad de ese blanco, que las moléculas tengan un comportamiento en el organismo que les permita alcanzar las concentraciones apropiadas en el lugar de acción, y que dicha molécula no sea tóxica. La información que nos permite concluir que un fármaco es seguro y eficaz se recolecta a través de estudios de laboratorio (preclínicos) y estudios en pacientes (clínicos) que toman años para ser completados. A lo largo de este extenso proceso se captura un gran volumen de información, mediante el trabajo de múltiples equipos de desarrollo experimentados, por medio de técnicas sofisticadas; sin embargo, existe una alta tasa de fallo de los estudios clínicos (1,6), porque muchas moléculas candidatas no llegan a ser eficaces o seguras en los pacientes. La falla de los estudios disminuye la eficiencia del proceso de desarrollo de nuevos fármacos, dada la alta inversión en dinero y tiempo de realizar estudios preclínicos y clínicos. Los modelos predictivos que resultan de algoritmos bien estructurados, elaborados por medio de técnicas de inteligencia artificial, podrían aumentar la tasa de efectividad de los estudios clínicos, al mejorar el proceso de identificación, priorización y validación de moléculas y blancos terapéuticos. Es interesante que cada vez más modelos de inteligencia artificial son utilizados en la investigación farmacéutica. La exploración de artículos publicados en el área de la salud, y que pueden ser encontrados en Pubmed, muestra que alrededor del 18% de los 165.992 artículos publicados en inteligencia artificial en salud tratan temas de farmacología (Figura 1).
A continuación, esta revisión se centrará en dos importantes aplicaciones de la inteligencia artificial en la farmacología. Primero se discutirá el papel de la inteligencia artificial en la investigación preclínica, específicamente en la biofarmacéutica y la química computacional para la identificación y validación de blancos terapéuticos y fármacos candidatos. Por último, se analizará el papel de la inteligencia artificial en la farmacología clínica, para la reutilización de fármacos y la selección de pacientes para los estudios clínicos.
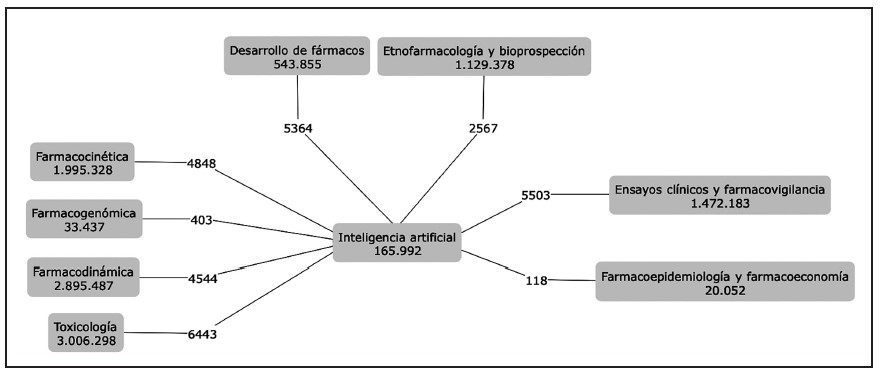
Figura 1. Exploración de artículos publicados en las áreas de la salud y la inteligencia artificial. El diagrama muestra categorías (texto) y volúmenes de publicaciones (números) en cada uno de los temas explorados. El sondeo de publicaciones en pubmed se realizó por medio de términos libres y términos MeSH para cada categoría. Los valores en las líneas conectoras muestran el número de artículos que existen en la intersección de la categoría específica y el área de la inteligencia artificial (datos de los autores).
Modelos de predicción computacional
La industria farmacéutica se puede beneficiar de la implementación de nuevas tecnologías, como la inteligencia artificial, para agilizar los procesos de descubrimiento y desarrollo de fármacos (Figura 2). La combinación entre inteligencia artificial y farmacología puede potenciar el desarrollo de medicamentos para patologías de alta complejidad, puesto que se pueden llegar a identificar mejores blancos terapéuticos, que están localizados en medio de vías de señalización, y redes de interacción entre proteínas complejas. El uso de inteligencia artificial puede además mejorar el tamizaje de compuestos con actividad farmacológica, aprovechar la información que proviene de estudios ómicos (genómica, transcriptómica, proteómica, metabolómica y otros) que generan grandes volúmenes de datos y seleccionar de manera más precisa los sujetos que podrían beneficiarse de un tratamiento de acuerdo con sus características biológicas (7,8). A pesar de la buena penetración de la inteligencia artificial en el campo de la farmacología (Figura 1), todavía se evidencian algunas falencias en cuanto a la disponibilidad, organización, heterogeneidad y calidad de los datos requeridos para generar mejores modelos de predicción (7,9).Aplicaciones de la inteligencia
artificial en el descubrimiento de
fármacos y la farmacología básica
Descubrimiento de compuestos
farmacéuticos
El procedimiento de desarrollo de un fármaco cuesta
alrededor de 2.6 billones de dólares y necesita aproximadamente 12 años
para ser cumplido (4). Este
proceso se puede dividir en cuatro fases: la selección
y validación del blanco terapéutico, la identificación
sistemática de compuestos con potencial farmacológico, la optimización
de las moléculas identificadas y la
evaluación preclínica y clínica de la seguridad y eficacia de los
fármacos (4). En los últimos años, con el
desarrollo de aplicaciones de la inteligencia artificial
en el proceso de descubrimiento de fármacos, se han
encontrado maneras de hacer que los procesos de desarrollo sean más
eficientes en tiempo y costo (4).
Los programas soportados en inteligencia artificial permiten la automatización de tareas complejas y repetitivas que normalmente requieren mucho tiempo. En los primeros pasos del descubrimiento de medicinas, esta automatización ha facilitado el análisis de ensayos celulares, la modelación de estructuras moleculares y sus interacciones, la planeación de la síntesis química de nuevas moléculas, la predicción de propiedades fisicoquímicas de compuestos y una gran variedad adicional de aplicaciones (1,2,6). Es muy útil en los primeros pasos del proceso del descubrimiento de medicinas generar modelos computacionales de las estructuras macromoleculares para las cuales se investigan compuestos moduladores. Un ejemplo de estas aplicaciones se encuentra en el modelamiento de proteínas.
El modelamiento de proteínas es muy importante para el descubrimiento de nuevas medicinas porque mucho del trabajo computacional está basado en modelar las interacciones entre ligandos potenciales y la estructura de la proteína. Una técnica bastante usada con este fin es el modelamiento por homología (del inglés homology modelling), por medio de la cual, ante la ausencia de resultados experimentales por métodos como la cristalografía de rayos X, es posible crear un modelo virtual de la proteína de interés a partir de la estructura ya conocida de otra proteína similar y que tiene una homología mayor al 30% (10). El modelamiento por homología suele ser problemático cuando no se cuenta con la estructura de proteínas similares y por esta razón es muy útil tener métodos de novo para modelar proteínas con base en la estructura de otras proteínas conocidas, pero no relacionadas. Con este fin es posible utilizar técnicas de inteligencia artificial a través el algoritmo AlphaFold (11) que predice, con una alta precisión, la estructura de una proteína. El algoritmo está entrenado con una base de datos de estructuras de proteínas determinadas experimentalmente. Con base en las distancias y ángulos de esas proteínas, y la secuencia de aminoácidos de la proteína de la cual queremos determinar la estructura, se genera un modelo tridimensional de la molécula objetivo con alta precisión.
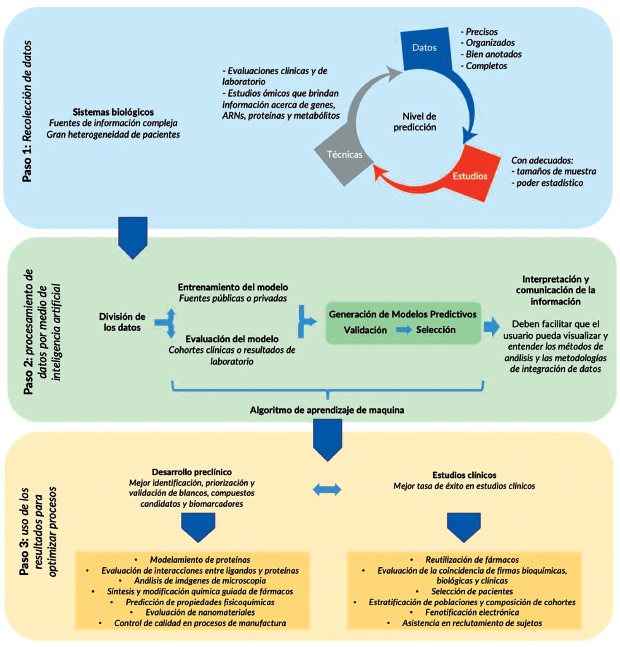
Figura 2. Resumen de aplicaciones de la inteligencia artificial en la farmacología. El proceso de desarrollo de fármacos (paso 1) inicia con la recolección de datos a partir de estudios de investigación preclínica y clínica. La calidad de los datos, recolectados a partir de técnicas precisas que hacen parte de la metodología de estudios bien diseñados, aumenta el nivel de predicción de los resultados, para orientar el desarrollo de fármacos. Los grandes volúmenes, variedad, y velocidad de los datos biológicos (las Vs de la ciencia de datos) recolectados en la investigación, dificultan que la información pueda ser procesada por métodos tradicionales. Sin embargo, estos datos pueden ser usados para entrenar y evaluar modelos predictivos, por medio de algoritmos de inteligencia artificial y técnicas de aprendizaje de maquina. La validación y selección de modelos robustos puede mejorar la tasa de éxito de procesos de investigación preclínica y clínica, por medio de la identificación, priorización y selección de mejores, más seguros y eficaces, fármacos candidatos. Los modelos de inteligencia artificial también pueden mejorar la estratificación y selección de poblaciones y pacientes para estudios clínicos. Finalmente, la inteligencia artificial puede permitir la reutilización de fármacos seguros, que no mostraron suficiente eficacia para una patología, pero que podrían ser útiles para el tratamiento de otras enfermedades.
La inteligencia artificial también ha sido aplicada en la predicción de interacciones entre proteínas y ligandos. Predecir estas interacciones es una herramienta muy útil para guiar modificaciones químicas en los ligandos y generar moléculas con un potencial más alto de ser medicamentos. Para predecir interacciones entre proteínas y sus ligandos es muy útil usar métodos de mecánica quántica (MQ) o métodos mezclados de mecánica quántica y mecánica molecular (MQ/MM) (12,13).
En estas técnicas se usa la mecánica quántica como método para poder calcular la densidad de electrones en todo el sistema (MQ) o sólo en una parte (MM/ MQ). Este cálculo puede ser usado para determinar la energía potencial de un sistema y, por extensión, la mejor forma en la cual un ligando se une a una proteína. Los cálculos de mecánica cuántica requieren mucho tiempo y son muy complicados. La inteligencia artificial puede disminuir de forma notoria el tiempo requerido para realizar estos cálculos (12,13). Sistemas de redes neuronales, que están entrenadas con datos de potenciales calculados usando mecánica cuántica, pueden predecir el potencial de una molécula mucho más rápido que cálculos ab inicio. Este tipo de cálculos requieren sólo las posiciones de los átomos del sistema investigado para poder predecir interacciones entre una macromolécula biológica y un ligando (14).
Respeto a la aplicación de la inteligencia artificial en el análisis de ensayos celulares, se ha demostrado que esa tecnología es muy eficiente en identificar y diferenciar imágenes. Una de las tareas menos eficientes en el proceso de descubrimiento de fármacos es analizar datos microscópicos de ensayos celulares. Estos ensayos se usan para investigar los efectos de fármacos potenciales sobre células in vitro en escalas muy grandes, donde cientos de miles de moléculas distintas se ensayan en paralelo (high-throughput screening) generando una gran cantidad de datos. Tradicionalmente se usan métodos de espectroscopia de fluorescencia u otro tipo de marcador fisicoquímico para leer los resultados de estos ensayos (15). La aplicación de la inteligencia artificial permite analizar, de forma masiva, pequeñas diferencias en las estructuras celulares a partir de imágenes de microscopía. Esta aplicación ha permitido diferenciar entre células de cáncer de mama y células no cancerígenas (16), y analizar automáticamente el tipo de célula con base en datos visuales como la localización de proteínas y contactos entre células (17).
En el proceso de descubrimiento de fármacos, después de haber identificado un compuesto, es necesario sintetizarlo. Este proceso puede ser muy lento porque requiere mucho trabajo en probar diferentes rutas de síntesis que podrían fallar. Debido a la inmensa cantidad de reacciones químicas que han sido publicadas, se han creado redes neuronales con la capacidad de diseñar rutas sintéticas útiles (18,19). Esas redes usan estrategias retrosintéticas como las que realizaría una persona experta en química. La retrosíntesis es una herramienta de la química orgánica donde a partir de la molécula deseada se proponen pasos sintéticos reversos hasta que se llega a moléculas más simples. Se han creado redes neuronales que pueden hacer este trabajo basados en reacciones químicas conocidas (18). La retrosíntesis es un procedimiento que requiere que la red neuronal sea capaz de reconocer lugares de la estructura donde se pueden inducir cambios e identificar cuales cambios serían los más adecuados. Para este tipo de algoritmo se usa el árbol de búsqueda de Monte Carlo (Monte Carlo tree search, MCTS). Una red neuronal basada en este algoritmo ha mostrado resultados efectivos y rápidos (18).
Determinar una ruta sintética no es el único problema que se encuentra al intentar sintetizar un ligando. Poder predecir el producto y el rendimiento de una reacción química muchas veces no resulta tan sencillo. Para resolver este problema también se ha aplicado la inteligencia artificial (20-24), y un ejemplo de su uso es el diseño de redes neuronales que pueden identificar el tipo de reacción (21) y pueden predecir el producto de una reacción (20,22). También se han reportado casos de aplicaciones de inteligencia artificial en donde se pudo entrenar una red neuronal para predecir los productos de reacciones de tipo acoplamiento de Buchwald-Hartwig, un tipo de reacción que es común en la química farmacéutica. Esta red neuronal fue entrenada sobre datos de química cuántica, como los momentos dipolares y las frecuencias vibracionales de los reactantes. Estos datos fueron calculados teóricamente usando química cuántica y sirvieron como descriptores de los reactantes. El rendimiento de las reacciones fue determinado experimentalmente haciendo varias reacciones en un rango nanomolar, usando un robot. La red neuronal creada fue capaz de determinar el rendimiento de las reacciones, después de haber sido entrenada sólo con base a los cálculos de los atributos cuánticos (20,22). Estas aplicaciones de la inteligencia artificial ayudan a hacer el trabajo de síntesis química mucho más fácil y rápido, y así se logra aumentar la velocidad del descubrimiento de nuevos compuestos farmacéuticos.
La inteligencia artificial también se ha usado para descubrir nuevas reacciones. Se han creado redes neuronales asociadas con robots que pueden probar varias combinaciones de reactivos para encontrar reacciones nuevas y reproducibles (20,21,25,26). Los robots prueban diferentes combinaciones de reactivos y usando una cámara (20,25), resonancia magnética nuclear y espectrometría infrarroja (27), pueden determinar la presencia de un nuevo producto. Este tipo de robots fueron usados para descubrir nuevas reacciones que pueden ser replicadas manualmente (28).
Después de haber identificado un ligando con actividad biológica, se generan compuestos similares para estudiar las partes de la molécula que son responsables de las propiedades farmacológicas. Una manera de asociar la estructura con la actividad son los estudios cuantitativos de la relación entre estructura y actividad (quantitative structure activity relationship studies, QSAR). Para estos estudios se usan pares de moléculas que se diferencian entre ellas sólo por un cambio en su estructura (matched molecular pair). Para este tipo de análisis se define un núcleo estructural de la molécula y se generan variantes con pequeños cambios (23,29). Una persona experta puede identificar patrones en pequeños grupos de moléculas, pero para grupos muy grandes se requieren algoritmos que realicen este trabajo de manera automática. De estos algoritmos existen unos de aprendizaje supervisado que son muy efectivos, aunque no generan asociaciones entre estructura y actividad inesperada tan efectivamente como los de aprendizaje no supervisado (29). También se ha logrado generar redes neuronales profundas que pueden tomar información de moléculas e identificar las partes estructurales más importantes para la actividad de estos compuestos (30-32). Estos algoritmos utilizan medidas conocidas de actividad biológica, de la mano de datos generados por el tamizaje virtual de grandes colecciones de compuestos sobre modelos virtuales de proteínas (33), datos exclusivos de estructura de las moléculas (31) o datos combinados de estructura de las moléculas y la estructura del sitio de unión en la proteína (32).
La inteligencia artificial también ha sido aplicada en la predicción de propiedades fisicoquímicas. Conocer estas propiedades es importante en el descubrimiento de nuevas medicinas porque pueden predecir, al menos en parte, el comportamiento biológico de una medicina.
Propiedades como el punto de fusión o el coeficiente de partición en agua/octanol (clogP) de un compuesto dependen de su estrutura molecular y afectan la biodisponibilidad de los fármacos (34). El punto de fusión es importante porque es una medida que se usa para predecir la solubilidad de un compuesto. El coeficiente de partición entre el agua y el octanol es importante para predecir la absorción de un compuesto (34). Para poder extraer los datos estructurales, de manera que puedan ser procesados por un software de inteligencia artificial, se usan métodos de abstracción de elementos estructurales como las posiciones, las cargas parciales y la conectividad de los átomos en una molécula, por medio de objetos computacionales como matrices de Coulomb, sistemas simplificados de entrada lineal (simplified molecular input line-entry system - SMILES), medidas de energía potencial y otros (35,36). Estos objetos, abstraídos de moléculas con propiedades fisicoquímicas conocidas, pueden ser usados para entrenar una red neuronal profunda, la cual podrá ser usada para predecir propiedades de otras moléculas. Metodologías así se han usado para predecir el punto de fusión (37) y el clogP (38).
La inteligencia artificial ha sido aplicada no sólo en los primeros pasos del proceso del descubrimiento de fármacos, sino también en otras áreas como la optimización de la administración de medicamentos. La tecnología farmacéutica ha logrado mejorar la farmacocinética de compuestos para el tratamiento de enfermedades como el cáncer. Para esto se han usado nanomateriales como nanotubos de carbono o nanopartículas (9), los cuales son muy grandes comparados con el tamaño de moléculas o péptidos que se usan como medicamentos. Los esfuerzos computacionales para caracterizar estos nanomateriales están basados más que todo en simulaciones por técnicas como simulaciones de dinámicas moleculares, que requieren mucho tiempo y poder computacional (9). Estos métodos simulan el movimiento y las interacciones de estos nanomateriales y permiten inferir propiedades como la capacidad de inserción en una membrana. Por el tamaño y la complejidad estructural de los nanomateriales es muy difícil generar una relación entre estructura y actividad cuantitativa, como se hace para medicamentos unimoleculares (39). Por esta razón, en el campo de la inteligencia artificial se usan descriptores de nanomateriales como los grupos químicos de su superficie y, en ocasiones, medidas experimentales como el tamaño o datos de actividad biológica. Este tipo de información ha sido usada por redes neuronales para poder predecir la nanohidrofobicidad de los nanomateriales, una medida asociada con su absorción (40).
Finalmente, uno de los últimos pasos en la fabricación de medicamentos es el control de calidad. La generación de varios lotes de medicamentos requiere el control de cada uno para comprobar una alta calidad de producto. Estos controles se hacen usando ensayos de disolución (41), química analítica y física. Estas mediciones generan un gran volumen de datos que pueden ser usado por redes neuronales para mejorar los procesos de fabricación. Se han usado redes neuronales para monitorear y predecir el perfil de disolución de lotes de fármacos (41). También se ha implementado la inteligencia artificial para monitorear otros procesos de fabricación, como la liofilización (42).
Aplicaciones de la inteligencia artificial en la farmacología clínica
La reutilización o reposicionamiento de medicamentos
A pesar de los grandes avances en la tecnología y en el conocimiento de la enfermedad humana, la traducción de estos beneficios en avances terapéuticos ha sido, hasta ahora, más lenta de lo esperada (43,44). Como se ha mencionado previamente, los desafíos que enfrenta la industria farmacéutica en el desarrollo de nuevos medicamentos son múltiples e incluyen una elevada tasa de fallo de los estudios clínicos (45,46), un tiempo prolongado para que llevar nuevos medicamentos al mercado, y cambios constantes en los requerimientos de las agencias regulatorias. Estos factores contribuyen a que el proceso de desarrollo de un nuevo medicamento tenga un costo elevado y que el retorno monetario en Investigación y Desarrollo (I&D) de la industria farmacéutica, pueda ser menor a la inversión (47). Estos elementos hacen que la industria farmacéutica pierda atractivo para la comunidad inversionista.La reutilización de medicamentos (también llamado reposicionamiento o reperfilado de medicamentos) es una estrategia que busca encontrar nuevos usos para medicamentos aprobados o en fase de investigación, que se encuentran fuera del alcance de la indicación para la cual fueron originalmente diseñados. Esta estrategia ofrece diversas ventajas sobre el desarrollo tradicional de un nuevo medicamento. La primera de ellas y quizás la más importante es la minimización del riesgo de fracaso: considerando que el medicamento reutilizado ya ha probado ser lo suficientemente seguro en modelos preclínicos y en humanos, es más probable que mantenga su perfil de seguridad y tolerabilidad en las indicaciones adicionales y, así, no haya un fracaso desde el punto de vista de seguridad en ensayos clínicos subsecuentes. Otra ventaja consiste en que el marco de tiempo para el desarrollo de medicamentos puede ser menor, dado que la mayoría del desarrollo preclínico, las evaluaciones de seguridad y, en algunos casos, el desarrollo de la formulación ya ha sido completado. Finalmente y como consecuencia de las anteriores, es altamente probable que se requiera menos inversión para la reutilización de un medicamento, en especial derivado de optimización de los estudios preclínicos y de fase I y II, aunque esto va a variar ampliamente dependiendo de la fase y del proceso de desarrollo del candidato a ser reutilizado (48).
En conjunto, todas estas ventajas derivadas de la estrategia de reutilización tienen el potencial de hacer el proceso de desarrollo de un nuevo medicamento menos riesgoso, con un retorno de la inversión más rápido y con menos costos asociados una vez se han contabilizado los fracasos. Se calcula que los costos derivados de traer un medicamento reutilizado al mercado son de 300 millones de dólares en promedio, comparado con el estimado, mencionado anteriormente, de ~2 a 3 billones de dólares requeridos para una nueva entidad química (49). Finalmente, los medicamentos reutilizados tienen el potencial de revelar nuevos blancos y vías terapéuticas que pueden ser explotadas.
Históricamente, el proceso de reutilización de medicamentos ha sido, en su gran mayoría, resultado de hechos oportunistas y fortuitos. De hecho, varios de los ejemplos más exitosos de reutilización de medicamentos no han involucrado un proceso de abordaje sistemático y han sido, por el contrario, fruto del azar. Dos ejemplos clásicos son el reposicionamiento de sildenafilo citrato, molécula originalmente desarrollada por la farmacéutica Pfizer como un medicamento antihipertensivo, que fue posteriormente reutilizado para el manejo de la disfunción eréctil con base a la experiencia clínica retrospectiva. El sildenafilo, comercializado posteriormente bajo la marca Viagra®, alcanzó una participación en el mercado de la disfunción eréctil de 47% y ventas globales de $2.05 billones de dólares, en el año 2012 (50). Otro ejemplo destacado es el reposicionamiento de talidomida, medicamento sedante originalmente utilizado para el manejo sintomático de náuseas y vómito asociados al embarazo, que fue retirado del mercado en 1961 debido a su asociación con defectos esqueléticos severos en niños nacidos de mujeres que tomaron el medicamento durante el primer trimestre de embarazo (43). Sin embargo, años más tarde y de manera fortuita, fue encontrada su eficacia para el manejo de eritema nodoso leproso (1964) y, más adelante, para el manejo de mieloma múltiple. Estas nuevas indicaciones llevaron al desarrollo y aprobación de un derivado de la talidomida aún más exitoso, la lenalidomida, medicamento que reportó ventas globales de $8.2 millones de dólares en 2017 (51). Ejemplos exitosos como los descritos han estimulado el desarrollo de abordajes sistemáticos para identificar compuestos candidatos a ser reutilizados, en especial para enfermedades raras, en las cuales el proceso de reutilización es, en muchas ocasiones, la única ruta para el desarrollo de medicamentos.
Típicamente, una estrategia de reutilización de un medicamento consiste en un proceso de tres pasos, previos a la decisión de llevar el medicamento a una fase de desarrollo clínico avanzado: Paso 1: evaluación hipotética del efecto del medicamento en modelos preclínicos para la identificación de un candidato adecuado para una indicación de interés; Paso 2: evaluación teórica del efecto del medicamento en estudios preclínicos y, Paso 3: evaluación de la eficacia en estudios clínicos fase II. De estos, el paso 1 es el más crítico y es donde los abordajes modernos de generación de hipótesis tienen una mayor utilidad. Estos abordajes sistemáticos pueden ser subdivididos a su vez en computacionales y experimentales.
Abordajes computacionales para la reutilización de medicamentos
La reutilización de fármacos requiere el análisis sistemático de fuentes de información de cualquier tipo (como expresión genética, estructura química, genotipo, información proteómica o información de historias clínicas), que permita la formulación de hipótesis de reutilización (52). Uno de los abordajes computacionales más utilizados con este propósito es el estudio de la coincidencia de firmas (del inglés signature matching). Este abordaje se basa en la comparación del conjunto de características únicas o la “firma” de un medicamento comparado con el de otro medicamento, enfermedad o fenotipo clínico (53,54). La firma de un medicamento puede derivarse de diferentes fuentes de información: datos de expresión y función de genes y proteínas (obtenidos por medio de transcriptómica, proteómica, metabolómica y estudios bioquímicos específicos), estructuras químicas o perfil de eventos adversos.La coincidencia de firmas transcriptómicas puede ser usada para hacer comparaciones entre medicamento y enfermedad (55) y entre medicamento y medicamento (56). En el primer caso, la firma transcriptómica de un medicamento en particular se obtiene de comparar el perfil de expresión genética de una muestra biológica antes y después de exponerse a un fármaco. La firma de expresión genómica diferencial resultante es después comparada con la del perfil de expresión genómica asociado a una enfermedad. El grado de correlación negativa entre la firma de expresión genómica del medicamento y el de la enfermedad (los genes regulados a la alta en la condición de enfermedad, que son a su vez regulados a la baja al exponerse al medicamento) puede permitir inferir un potencial efecto del medicamento en la enfermedad (57,58). Este abordaje computacional se basa en el Principio de Reversión de Firma (SRP por sus siglas en inglés, Signature Reversion Principle), el cual asume que un medicamento tiene la capacidad de revertir el patrón de expresión de un conjunto de genes que son característicos del fenotipo particular de una enfermedad y, de esta manera, revertir el fenotipo en sí mismo. Este principio ha sido empleado ampliamente para identificar medicamentos que podrían ser reposicionados como sensibilizadores a la quimioterapia, tomando como base el perfil de firma de resistencia a los medicamentos contra el cáncer (59).
Los abordajes de similitud entre medicamentos buscan identificar mecanismos de acción similares en medicamentos de otra manera disímiles, bien sea por que pertenecen a diferentes clases terapéuticas o son estructuralmente diferentes. Este principio es llamado “culpable por asociación” (del inglés guilty by assotiation) (60) y es de utilidad en la identificación de blancos alternativos para medicamentos existentes, o potenciales efectos off-target que pueden ser investigados para aplicaciones clínicas futuras (53).
El segundo tipo de proceso de coincidencia de firmas usado en el reposicionamiento de medicamentos se basa en las estructuras químicas y su relación con la actividad biológica. Este proceso se fundamenta en que, al encontrar similitudes en la estructura química de dos medicamentos, se puede inferir una actividad biológica similar (61).
Finalmente, existe el proceso de la coincidencia de firmas de eventos adversos de medicamentos. Esta alternativa se basa en la hipótesis de que dos medicamentos con un perfil de eventos adversos similar pueden presentar una actividad en un mismo blanco terapéutico (proteína o vía de señalización) (55).
Inteligencia artificial y Machine Learning (ML) en el reposicionamiento de medicamentos
Los abordajes de coincidencia de firmas involucran el análisis de un masivo y rápidamente creciente volumen de información proveniente de estudios “ómicos”, lo cual genera el desafío de identificar coincidencias obvias y no obvias entre las diferentes firmas, y es justo en este punto donde la inteligencia artificial desempeña un papel determinante. Los métodos de ML para el abordaje del reposicionamiento de medicamentos se pueden dividir en tres categorías: métodos basados en redes, minería de texto y abordaje semántico.- Los métodos basados en redes se dividen a su vez en métodos de racimo y abordaje de propagación. En el primer método, la búsqueda de posibles asociaciones entre medicamentos, enfermedades y dianas putativas se realiza en pequeños racimos embebidos dentro de una red más amplia. El método del abordaje de propagación se basa en el flujo de un conocimiento previamente adquirido a través de diferentes capas de una red. Según la manera en la que sea tratada esta red, este método puede presentar un abordaje local (abordaje de segmentos de la red) o un abordaje global (abordaje de la red como un todo) (62). Adicionalmente, si la composición de la red creada usa un único tipo de información, como interacción entre proteínas, se caracteriza como homogénea; mientras que si la red creada involucra diferentes tipos de información, se conoce como heterogénea (63)
- En el método de minería de texto, el proceso involucra la búsqueda de una vasta cantidad de literatura disponible sobre los términos de interés y el filtro de las fuentes para extraer la información relevante. En lo referente al reposicionamiento de medicamentos, el método de referencia es descrito como el “ABC”, el cual se basa en el concepto de que si A se relaciona con B, y B con C, es entonces es posible una conexión entre A y C (64).
- En el método basado en semántica, la creación de redes es guiada por el conocimiento biomédico previo, el cual es utilizado para identificar nuevas interacciones y relaciones que se encuentren presentes en estas redes (65).
Inteligencia artificial en el diseño y conducción de estudios clínicos
Como se discutió previamente, una de las barreras más pronunciadas en el desarrollo del portafolio de nuevos medicamentos es la elevada tasa de fracaso de los ensayos clínicos, lo cual se evidencia en que menos de un tercio de los compuestos en fase II avanzan a fase III (66) y más de un tercio de los compuestos en fase III no avanzan hacia la aprobación regulatoria y comercialización (67). Los factores más críticos que ocasionan que un ensayo clínico no sea exitoso son los mecanismos de selección de la cohorte de pacientes y de reclutamiento, los cuales fallan en incorporar al estudio los pacientes más adecuados de manera oportuna. Otra limitación importante es la falta de infraestructura técnica para administrar y conducir estudios clínicos, sumado a la ausencia de un sistema de control confiable y eficiente de adherencia y monitoreo de pacientes, y detección de desenlaces clínicos.La inteligencia artificial representa una herramienta de mucha utilidad para abordar los desafíos actuales en el diseño de estudios clínicos. A través de técnicas de aprendizaje de máquina (ML) y aprendizaje profundo (Deep Learning - DL) es posible identificar patrones de significancia en grandes bases de datos compuestas por texto, imágenes o palabras habladas. A través del Procesamiento del Lenguaje Natural (PLN) es posible comprender y correlacionar contenido en lenguaje escrito o hablado, y las Interfaces de Hombre-Máquina (IHM) permiten el intercambio natural de información entre humanos y computadores. Estas herramientas pueden ser usadas para correlacionar amplias y diversas fuentes de información como historias clínicas electrónicas, literatura médica y bases de información de estudios clínicos, para mejorar la coincidencia entre pacientes y estudios y el reclutamiento previo al inicio del estudio. Estas herramientas también permiten monitorear pacientes de manera automática y continua durante el estudio clínico, optimizando el control de la adherencia y la evaluación de desenlaces clínicos para lograr conducir estudios clínicos con una mayor confiabilidad y eficiencia.
Selección de pacientes
Uno de los mayores retos al conducir un ensayo clínico es la selección y el reclutamiento de pacientes que cumplan con los criterios de elegibilidad, idoneidad, motivación y empoderamiento para ser vinculados al estudio. Las demoras en la selección y reclutamiento de pacientes constituye la causa número uno de retraso en los ensayos clínicos y se calcula que un porcentaje tan elevado como el 86% de los estudios clínicos no cumple con sus metas de tiempos de reclutamiento (68). Los sistemas conducidos por inteligencia artificial y ML pueden ser de mucha utilidad en la optimización de la composición de las cohortes de pacientes y proveer asistencia en el reclutamiento de pacientes.Composición de cohortes
En un mundo ideal la evaluación de la idoneidad de los sujetos a ser reclutados en un estudio clínico debería considerar desde el perfilamiento del genoma hasta el exposoma de cada paciente, para determinar si los biomarcadores en los cuales actúa el medicamento se encuentran suficientemente expresados en un paciente en particular (69). Para alcanzar esta visión, es necesario usar métodos de análisis sofisticados que combinan información de estudios “ómicos”, historias clínicas electrónicas y de otros datos del paciente (en diferentes formatos según su dueño y ubicación) para identificar biomarcadores. Estos biomarcadores pueden tener una utilidad diagnóstica, pronóstica o terapéutica, que mejora la evaluación de los desenlaces clínicos y, como consecuencia, ayudan a identificar y caracterizar apropiadamente las subpoblaciones de pacientes. Esta corresponde a una oportunidad única para el uso de PLN y algoritmos de visión de computador (como el reconocimiento óptico de caracteres), para automatizar la lectura y compilación de esas complejas fuentes de información. Mas aún, considerando el desafío que implica el tratamiento de información proveniente de diferentes fuentes y formatos como el de las historias clínicas electrónicas (debido a su volumen, velocidad, veracidad y variedad), la naturaleza agnóstica de los modelos de inteligencia artificial representa una herramienta única para la armonización de la información.De acuerdo con la Agencia de Alimentos y Medicamentos, (FDA por sus siglas en inglés, Food and Drug Agency), los modelos y métodos de inteligencia artificial pueden ser usados para optimizar la selección de una cohorte de pacientes a través de las siguientes estrategias: 1) reduciendo la heterogeneidad de la población, 2) a través de la elección de pacientes en los que sea más probable obtener un desenlace clínico medible (también llamado enriquecimiento de pronóstico) y 3) por medio de la identificación de una población que tenga mayor probabilidad de responder al tratamiento (también llamado enriquecimiento predictivo).
La fenotipificación electrónica es una disciplina ampliamente establecida en informática de salud que se enfoca en reducir la heterogeneidad de la población a través de la optimización del proceso de la identificación de pacientes con características de interés, que pueden ser tan sencillas como la presencia de diabetes mellitus tipo 2, o tan compleja como la presencia de cáncer de próstata estadio II con urgencia miccional y sin presencia de infección de vías urinarias. Si bien esta selección puede hacerse de manera tradicional seleccionando manualmente las historias clínicas, en la medida en que la información sea más voluminosa y compleja, las técnicas de inteligencia artificial han demostrado un destacado rendimiento administrando fuentes de información del mundo real (70).
A pesar de la innegable utilidad de la fenotipificación electrónica en reducir la heterogeneidad de la población de pacientes, esta técnica no está diseñada para alcanzar un enriquecimiento pronóstico o predictivo, el cual requiere de modelos más complejos necesarios para caracterizar y evaluar la progresión de la enfermedad. Existen algunos ejemplos del uso de métodos de ML en patologías neurológicas donde se combinan diferentes mediciones que proveen información pronóstica (71, 72). Actualmente se encuentran en desarrollo diferentes métodos de ML para el modelamiento de la progresión de la enfermedad, que buscan incrementar su precisión, particularmente en enfermedades neurodegenerativas (73-76).
Asistencia en el reclutamiento
La complejidad de los criterios de elegibilidad para un estudio clínico, tanto en el número de criterios como en el lenguaje médico usado, hacen que para los pacientes resulte complejo comprender y evaluar su propia elegibilidad. Una solución convencional es la extracción manual de información de interés a partir de las historias clínicas, lo cual implica un proceso prolongado y con una elevada carga para los investigadores. Existen múltiples técnicas de IA que pueden ofrecer asistencia valiosa en encontrar las “agujas en el pajar” de las historias clínicas. Las técnicas de PLN pueden ser usadas para comprender el lenguaje escrito y hablado que está presente en una variedad de tipos de información estructurada y no estructurada de las historias clínicas (77). Las técnicas de razonamiento permiten que un contenido denso sea convertido en recomendaciones accionables para el humano tomador de decisiones (78). Métodos de ML y, en particular, el aprendizaje de refuerzo profundo, empoderan los sistemas para aprender e integrar la retroalimentación en la calidad de su resultado analítico y en integrarlo en algoritmos (79).Conclusión
Un gran desafío de la medicina moderna es la gestión de los grandes volúmenes de información que se producen a partir del trabajo en investigación y la atención en salud de los pacientes y comunidades. El reto por lo tanto es el desarrollo de herramientas que permitan administrar ese masivo volumen de información para tomar decisiones adecuadas en salud, entre ellas, el desarrollo de nuevos medicamentos que sean eficaces para prevenir, controlar y curar enfermedades con crecientes necesidades insatisfechas. Las diferentes aplicaciones de la inteligencia artificial representan una herramienta de inmensa utilidad para analizar e interpretar ese masivo volumen de datos y permitir a los investigadores desarrollar medicamentos con mayor velocidad y costo-eficiencia. Por lo tanto es crítico que los sistemas educativos y productivos en el área de la salud, ofrezcan oportunidades de formación y uso de inteligencia artificial que cierren la brecha de habilidades y conocimiento técnico en el personal de salud.Referencias
1.
Zhavoronkov A, Vanhaelen Q, Oprea TI. Will Artificial Intelligence for
Drug Discovery Impact Clinical Pharmacology? Clin Pharmacol Ther.
2020;107 (4):780-5.2. Paul D, Sanap G, Shenoy S, Kalyane D, Kalia K, Tekade RK. Artificial intelligence in drug discovery and development. Drug Discov Today. 2021;26 (1):80-93.
3. Yang X, Wang Y, Byrne R, Schneider G, Yang S. Concepts of Artificial Intelligence for Computer-Assisted Drug Discovery. Chem Rev. 2019;119 (18):10520-94.
4. Chan HCS, Shan H, Dahoun T, Vogel H, Yuan S. Advancing Drug Discovery via Artificial Intelligence. Trends Pharmacol Sci. 2019;40 (10):801.
5. Silcox C. La inteligencia artificial en el sector salud. Banco Interamericano de Desarrollo; 2020.
6. Vamathevan J, Clark D, Czodrowski P, Dunham I, Ferran E, Lee G, et al. Applications of machine learning in drug discovery and development. Nat Rev Drug Discov. 2019;18 (6):463-77.
7. Koromina M, Pandi MT, Patrinos GP. Rethinking Drug Repositioning and Development with Artificial Intelligence, Machine Learning, and Omics. OMICS. 2019;23 (11):539-48.
8. Savage N. Tapping into the drug discovery potential of AI. Biopharma Dealmakers [Internet]. 2021. Available from: https://www.nature.com/articles/d43747-021-00045-7.
9. Zhu H. Big Data and Artificial Intelligence Modeling for Drug Discovery. Annu Rev Pharmacol Toxicol. 2020;60:573-89.
10. Cavasotto CN, Phatak SS. Homology modeling in drug discovery: current trends and applications. Drug Discov Today. 2009 Jul;14(13-14):676-83. doi:10.1016/j.drudis.2009.04.006. Epub 2009 May 5.
11. Senior AW, Evans R, Jumper J, Kirkpatrick J, Sifre L, Green T, et al. Improved protein structure prediction using potentials from deep learning. Nature. 2020 Jan;577(7792):706-710. doi: 10.1038/s41586-019-1923-7. Epub 2020 Jan 15.
12. Hayik SA, Dunbrack R Jr, Merz KM Jr. A Mixed QM/MM Scoring Function to Predict Protein-Ligand Binding Affinity. J Chem Theory Comput. 2010 Sep 1;6(10):3079-3091. doi: 10.1021/ct100315g.
13. Wang M, Mei Y, Ryde U. Predicting Relative Binding Affinity Using Nonequilibrium QM/MM Simulations. J Chem Theory Comput. 2018 Dec 11;14(12):6613-6622. doi:10.1021/acs.jctc.8b00685. Epub 2018 Nov 8.
14. Zhang YJ, Khorshidi A, Kastlunger G, Peterson AA. The potential for machine learning in hybrid QM/MM calculations. J Chem Phys. 2018 Jun 28;148(24):241740. doi: 10.1063/1.5029879.
15. Sittampalam GS, Kahl SD, Janzen WP. High-throughput screening: advances in assay technologies. Curr Opin Chem Biol. 1997 Oct;1(3):384-91. doi: 10.1016/s1367- 5931(97)80078-6.
16. Ahuja A, Al-Zogbi L, Krieger A. Application of noisereduction techniques to machine learning algorithms for breast cancer tumor identification. Comput Biol Med. 2021 Aug;135:104576. doi: 10.1016/j.compbiomed.2021.104576. Epub 2021 Jun 19.
17. Nitta N, Sugimura T, Isozaki A, Mikami H, Hiraki K, Sakuma S, et al. Intelligent Image-Activated Cell Sorting. Cell. 2018 Sep 20;175(1):266-276.e13. doi: 10.1016/j. cell.2018.08.028. Epub 2018 Aug 27.
18. Segler MHS, Preuss M, Waller MP. Planning chemical syntheses with deep neural networks and symbolic AI. Nature. 2018 Mar 28;555(7698):604-610. doi: 10.1038/ nature25978.
19. Coley CW, Green WH, Jensen KF. Machine Learning in Computer-Aided Synthesis Planning. Acc Chem Res. 2018 May 15;51(5):1281-1289. doi: 10.1021/acs. accounts.8b00087. Epub 2018 May 1.
20. Maryasin B, Marquetand P, Maulide N. Machine Learning for Organic Synthesis: Are Robots Replacing Chemists? Angew Chem Int Ed Engl. 2018 Jun 11;57(24):6978- 6980. doi: 10.1002/anie.201803562. Epub 2018 Apr 27.
21. Wei JN, Duvenaud D, Aspuru-Guzik A. Neural Networks for the Prediction of Organic Chemistry Reactions. ACS Cent Sci. 2016 Oct 26;2(10):725-732. doi: 10.1021/acscentsci.6b00219. Epub 2016 Oct 14.
22. Ahneman DT, Estrada JG, Lin S, Dreher SD, Doyle AG. Predicting reaction performance in C-N crosscoupling using machine learning. Science. 2018 Apr 13;360(6385):186-190. doi: 10.1126/science.aar5169. Epub 2018 Feb 15. Erratum in: Science. 2018 Apr 13;360(6385).
23. Coley CW, Barzilay R, Jaakkola TS, Green WH, Jensen KF. Prediction of Organic Reaction Outcomes Using Machine Learning. ACS Cent Sci. 2017 May 24;3(5):434-443. doi: 10.1021/acscentsci.7b00064. Epub 2017 Apr 18.
24. Zhou Z, Li X, Zare RN. Optimizing Chemical Reactions with Deep Reinforcement Learning. ACS Cent Sci. 2017 Dec 27;3(12):1337-1344. doi: 10.1021/acscentsci.7b00492. Epub 2017 Dec 15.
25. Caramelli D, Salley D, Henson A, Camarasa GA, Sharabi S, Keenan G, et al. Networking chemical robots for reaction multitasking. Nat Commun. 2018 Aug 24;9(1):3406. doi: 10.1038/s41467-018-05828-8.
26. Granda JM, Donina L, Dragone V, Long DL, Cronin L. Controlling an organic synthesis robot with machine learning to search for new reactivity. Nature. 2018 Jul;559(7714):377-381. doi: 10.1038/s41586-018-0307-8. Epub 2018 Jul 18.
27. Wollenberg A, Flohr C, Simon D, Cork MJ, Thyssen JP, Bieber T, et al. European Task Force on Atopic Dermatitis statement on severe acute respiratory syndrome coronavirus 2 (SARS-Cov-2) infection and atopic dermatitis. J Eur Acad Dermatol Venereol. 2020;34 (6):e241-e2.
28. Perera D, Tucker JW, Brahmbhatt S, Helal CJ, Chong A, Farrell W, et al. A platform for automated nanomolescale reaction screening and micromole-scale synthesis in flow. Science. 2018 Jan 26;359(6374):429-434. doi: 10.1126/science.aap9112.
29. Tyrchan C, Evertsson E. Matched Molecular Pair Analysis in Short: Algorithms, Applications and Limitations. Comput Struct Biotechnol J. 2016 Dec 13;15:86-90. doi:10.1016/j.csbj.2016.12.003.PMID: 28066532; PMCID: PMC5198793.
30. Sheridan RP, Wang WM, Liaw A, Ma J, Gifford EM. Extreme Gradient Boosting as a Method for Quantitative Structure-Activity Relationships. J Chem Inf Model. 2016 Dec 27;56(12):2353-2360. doi: 10.1021/acs.jcim.6b00591. Epub 2016 Dec 13.
31. Sheridan RP, Wang WM, Liaw A, Ma J, Gifford EM. Extreme Gradient Boosting as a Method for Quantitative Structure-Activity Relationships. Journal of Chemical Information and Modeling2016. p. 2353-60.
32. Wallach I, Dzamba M, Heifets A. AtomNet: A Deep Convolutional Neural Network for Bioactivity Prediction in Structure-based Drug Discovery. 2015. p. 1-11.
33. Sampaio MF, Hirata MH, Hirata RD, Santos FC, Picciotti R, Luchessi AD, et al. AMI is associated with polymorphisms in the NOS3 and FGB but not in PAI-1 genes in young adults. Clin Chim Acta. 2007;377 (1-2):154-62.
34. Mao F, Kong Q, Ni W, Xu X, Ling D, Lu Z, et al. Melting Point Distribution Analysis of Globally Approved and Discontinued Drugs: A Research for Improving the Chance of Success of Drug Design and Discovery. ChemistryOpen. 2016 Mar 21;5(4):357-68. doi: 10.1002/open.201600015.
35. Sanchez-Lengeling B, Aspuru-Guzik A. Inverse molecular design using machine learning: Generative models for matter engineering. Science. 2018 Jul 27;361(6400):360-365. doi: 10.1126/science.aat2663. Epub 2018 Jul 26.
36. Shen J, Nicolaou CA. Molecular property prediction: recent trends in the era of artificial intelligence. Drug Discov Today Technol. 2019 Dec;32-33:29-36. doi: 0.1016/j.ddtec.2020.05.001. Epub 2020 Jul 1.
37. Sivaraman G, Jackson NE, Sanchez-Lengeling B, Vázquez-Mayagoitia Á, Aspuru-Guzik A, Vishwanath V, et al. A diversified machine learning strategy for predicting and understanding molecular melting points. ChemRxiv2019. p. 1-42.
38. Lenselink EB, Stouten PFW. Multitask machine learning models for predicting lipophilicity (logP) in the SAMPL7 challenge. J Comput Aided Mol Des. 2021 Aug;35(8):901-909. doi: 10.1007/s10822-021-00405-6. Epub 2021 Jul 17.
39. Puzyn T, Leszczynska D, Leszczynski J. Toward the development of "nano-QSARs": advances and challenges. Small. 2009 Nov;5(22):2494-509. doi: 10.1002/smll.200900179.
40. Li S, Zhai S, Liu Y, Zhou H, Wu J, Jiao Q, et al. Experimental modulation and computational model of nanohydrophobicity. Biomaterials. 2015 Jun;52:312-7. doi: 10.1016/j.biomaterials.2015.02.043. Epub 2015 Feb 28.
41. Krajišnik D, Stepanović-Petrović R, Tomić M, Micov A, Ibrić S, Milić J. Application of artificial neural networks in prediction of diclofenac sodium release from drug-modified zeolites physical mixtures and antiedematous activity assessment. J Pharm Sci. 2014 Apr;103(4):1085-94. doi: 10.1002/jps.23869. Epub 2014 Feb 4.
42. Drăgoi EN, Curteanu S, Fissore D. On the Use of Artificial Neural Networks to Monitor a Pharmaceutical Freeze-Drying Process. Drying Technology 31(1):72-81.
43. Ashburn TT, Thor KB. Drug repositioning: identifying and developing new uses for existing drugs. Nat Rev Drug Discov. 2004;3 (8):673-83.
44. Scannell JW, Blanckley A, Boldon H, Warrington B. Diagnosing the decline in pharmaceutical R&D efficiency. Nat Rev Drug Discov. 2012;11 (3):191-200.
45. Pammolli F, Magazzini L, Riccaboni M. The productivity crisis in pharmaceutical R&D. Nat Rev Drug Discov. 2015 Jul;14(7):475-86.
46. Waring MJ, Arrowsmith J, Leach AR, Leeson PD, Mandrell S, Owen RM, et al. An analysis of the attrition of drug candidates from four major pharmaceutical companies. Nat Rev Drug Discov. 2015;14 (7):475-86.
47. Pushpakom S, Iorio F, Eyers PA, Escott KJ, Hopper S, Wells A, et al. Drug repurposing: progress, challenges and recommendations. Nat Rev Drug Discov. 2019;18 (1):41-58.
48. Breckenridge A, Jacob R. Overcoming the legal and regulatory barriers to drug repurposing. Nat Rev Drug Discov. 2019;18 (1):1-2.
49. Nosengo N. Can you teach old drugs new tricks? Nature. 2016;534 (7607):314-6.
50. Pfizer’s Expiring Viagra Patent Adversely Affects Other Drugmakers Too. Forbes. 2013 December 20, 2013.
51. Urquhart L. Market watch: Top drugs and companies by sales in 2017. Nat Rev Drug Discov. 2018 Mar 28;17(4):232.
52. Hurle MR, Yang L, Xie Q, Rajpal DK, Sanseau P, Agarwal P. Computational drug repositioning: from data to therapeutics. Clin Pharmacol Ther. 2013 Apr;93(4):335-41.
53. Keiser MJ, Setola V, Irwin JJ, Laggner C, Abbas AI, Hufeisen SJ, et al. Predicting new molecular targets for known drugs. Nature. 2009;462 (7270):175-81.
54. Hieronymus H, Lamb J, Ross KN, Peng XP, Clement C, Rodina A, et al. Gene expression signature-based chemical genomic prediction identifies a novel class of HSP90 pathway modulators. Cancer Cell. 2006;10 (4):321-30.
55. Dudley JT, Deshpande T, Butte AJ. Exploiting drugdisease relationships for computational drug repositioning. Brief Bioinform. 2011 Jul;12(4):303-11.
56. Iorio F, Rittman T, Ge H, Menden M, Saez-Rodriguez J. Transcriptional data: a new gateway to drug repositioning? Drug Discov Today. 2013 Apr;18(7-8):350-7.
57. Dudley JT, Sirota M, Shenoy M, Pai RK, Roedder S, Chiang AP, et al. Computational repositioning of the anticonvulsant topiramate for inflammatory bowel disease. Sci Transl Med. 2011 Aug 17;3(96):96ra76.
58. Sirota M, Dudley JT, Kim J, Chiang AP, Morgan AA, Sweet-Cordero A, et al. Discovery and preclinical validation of drug indications using compendia of public gene expression data. Sci Transl Med. 2011 Aug 17;3(96):96ra77.
59. Wei G, Twomey D, Lamb J, Schlis K, Agarwal J, Stam RW, et al. Gene expression-based chemical genomics identifies rapamycin as a modulator of MCL1 and glucocorticoid resistance. Cancer Cell. 2006 Oct;10(4):331- 42.
60. Chiang AP, Butte AJ. Systematic evaluation of drug-disease relationships to identify leads for novel drug uses. Clin Pharmacol Ther. 2009 Nov;86(5):507-10.
61. Oprea TI, Tropsha A, Faulon JL, Rintoul MD. Systems chemical biology. Nat Chem Biol. 2007 Aug;3(8):447-50.
62. Emig D, Ivliev A, Pustovalova O, Lancashire L, Bureeva S, Nikolsky Y, et al. Drug target prediction and repositioning using an integrated network-based approach. PLoS One. 2013;8 (4):e60618.
63. Wu Z, Wang Y, Chen L. Network-based drug repositioning. Mol Biosyst. 2013 Jun;9(6):1268-81.
64. Weeber M, Klein H, De Jong-Van Den Berg L, Vos R. Using concepts in literature-based discovery: Simulating Swanson’s Raynaud–fish oil and migraine–magnesium discoveries. JASIST. 2001;52 (7):548-57.
65. Xue H, Li J, Xie H, Wang Y. Review of Drug Repositioning Approaches and Resources. Int J Biol Sci. 2018 Jul 13;14(10):1232-1244.
66. Hay M, Thomas DW, Craighead JL, Economides C, Rosenthal J. Clinical development success rates for investigational drugs. Nat Biotechnol. 2014 Jan;32(1):40-51.
67. Wong CH, Siah KW, Lo AW. Estimation of clinical trial success rates and related parameters. Biostatistics. 2019;20 (2):273-86.
68. CLINPAL. Recruitment infographic 2021 [Available from: https://www.clinpal.com/recruitment-infographic/.
69. Harrer S, editor Measuring life: sensors and analytics for precision medicine. SPIE Microtechnologies; 2015; Barcelona: Proc. SPIE 9518, Bio-MEMS and Medical Microdevices II, 951802.
70. Banda JM, Seneviratne M, Hernandez-Boussard T, Shah NH. Advances in Electronic Phenotyping: From Rule-Based Definitions to Machine Learning Models. Annu Rev Biomed Data Sci. 2018;1:53-68.
71. Goudey B, Fung BJ, Schieber C, Faux NG; Alzheimer’s Disease Metabolomics Consortium; Alzheimer’s Disease Neuroimaging Initiative. A blood-based signature of cerebrospinal fluid Aβ1-42 status. Sci Rep. 2019 Mar 11;9(1):4163. doi: 10.1038/s41598-018-37149-7.
72. Palmqvist S, Insel PS, Zetterberg H, Blennow K, Brix B, Stomrud E, et al. Accurate risk estimation of betaamyloid positivity to identify prodromal Alzheimer’s disease: Cross-validation study of practical algorithms. Alzheimers Dement. 2019 Feb;15(2):194-204.
73. Ghosh S, Sun Z, Li Y, Cheng Y, Mohan A, Sampaio C, et al. An Exploration of Latent Structure in Observational Huntington’s Disease Studies. AMIA Jt Summits Transl Sci Proc. 2017;2017:92-102.
74. Sun Z, Li Y, Ghosh S, Cheng Y, Mohan A, Sampaio C, et al. A Data-Driven Method for Generating Robust Symptom Onset Indicators in Huntington’s Disease Registry Data. AMIA Annu Symp Proc. 2017;2017:1635-44.
75. Sun Z, Ghosh S, Li Y, Cheng Y, Mohan A, Sampaio C, et al. A probabilistic disease progression modeling approach and its application to integrated Huntington’s disease observational data. JAMA Open. 2019;2 (1):123- 30.
76. Schobel SA, Palermo G, Auinger P, Long JD, Ma S, Khwaja OS, et al. Motor, cognitive, and functional declines contribute to a single progressive factor in early HD. Neurology. 2017 Dec 12;89(24):2495-2502.
77. Fogel DB. Factors associated with clinical trials that fail and opportunities for improving the likelihood of success: A review. Contemp Clin Trials Commun. 2018 Aug 7;11:156-164.
78. LeCun Y. The Power and Limits of Deep Learning: In his IRI Medal address, Yann LeCun maps the development of machine learning techniques and suggests what the future may hold. Res Technol Manag. 2018;61 (6):22-7.
79. LeCun Y, Bengio Y, Hinton G. Deep learning. Nature. 2015 May 28;521(7553):436-44.
Recibido: 12 de Noviembre, 2021
Aceptado: 22 de Noviembre, 2021
Correspondencia:
Ricardo A. Peña-Silva
rpena@uniandes.edu.co