Resúmen
El cáncer de mama (CM) es la segunda neoplasia más común a nivel
mundial y el cáncer más frecuente en mujeres. En los últimos años se
han logrado diversos avances en la detección temprana, tratamiento y
comprensión de la biología de esta enfermedad. La heterogeneidad
biológica del CM tiene
implicaciones tanto en el pronóstico, como en la toma de decisiones
terapéuticas.
Probablemente uno de los éxitos más importantes a principios del siglo
XX, fue la alteración del
entorno endócrino interno tumoral a través de la terapia hormonal. El
descubrimiento de la terapia
endocrina permitió pasar de tratamientos ablativos quirúrgicos y
quimioterapias agresivas, a administrar terapias dirigidas, mejor
toleradas y que mejoran la calidad de vida en mujeres con CM
hormonodependiente.
El fundamento de la terapia endocrina es la dependencia bien demostrada
de la mama a la estimulación hormonal. En los años 1970’s, el modulador
selectivo del receptor de estrógeno, tamoxifeno
emerge como una nueva terapia para mujeres con CM avanzado; en las
décadas subsecuentes, surgen
numerosas y diversas terapias endocrinas selectivas, como los agonistas
de la liberación de hormona
luteinizante y los inhibidores de aromatasa, como una estrategia
adicional en el tratamiento de esta
enfermedad. Estas terapias fueron estudiadas de manera inicial en el
escenario metastásico y posteriormente fueron adoptadas en el escenario
adyuvante, en mujeres con enfermedad temprana. De manera más reciente
se han descubierto los inhibidores de cinasa dependiente de ciclina,
que asociados a
la terapia hormonal han logrado una mejoría significativa en desenlaces
clínicos y tasas de respuestas.
En este artículo realizaremos una revisión cronológica de los
principales hitos en la evolución de la
terapia endocrina a lo largo de los últimos 100 años, destacando cada
clase de agente y los ensayos
claves que han llevado a cambios en la práctica moderna.
Palabras clave: Cáncer de mama; receptor de estrógenos;
tratamiento endócrino.

¹ Departamento de Hemato-Oncología, Instituto Nacional de Ciencias
Médicas y Nutrición Salvador Zubirán, Ciudad de México,
México.
² Departamento de Anatomía
Patológica, Instituto Nacional de
Ciencias Médicas y Nutrición Salvador Zubirán, Ciudad de México,
México.
³ Servicio de Geriatría,
Instituto Nacional de Ciencias Médicas y
Nutrición Salvador Zubirán, Ciudad de México, México.
HISTORY OF HORMONE RECEPTORS AND HORMONE
THERAPY IN BREAST CANCER
Abstract
Breast cancer (BC) is the second most common neoplasm worldwide and the
most common
cancer in women. Over the past few years, various advances have been
made in early detection, treatment, and understanding of the biology of
this disease. The biological heterogeneity
of BC has implications for both prognosis and therapeutic
decision-making.
Probably one of the most important successes at the beginning of the
20th century was the
alteration of the tumor’s internal endocrine environment through
hormonal therapy. The discovery of endocrine therapy made it possible
to move from surgical ablative treatments and
aggressive chemotherapies to the administration of targeted therapies
that are better tolerated
and that improve the quality of life of women with hormone-dependent
BC.
The rationale for endocrine therapy is the well-established dependence
of the breast to
hormonal stimulation. In the 1970s, the selective estrogen receptor
modulator, tamoxifen,
emerged as a new therapy for women with advanced BC; in subsequent
decades, numerous
and diverse selective endocrine therapies emerged, such as luteinizing
hormone agonists and
aromatase inhibitors, as an additional strategy in the treatment of
this disease. These therapies were initially studied in the metastatic
setting and have subsequently been adopted in the
adjuvant setting for women with early disease. More recently,
cyclin-dependent kinase inhibitors have been discovered and, associated
with hormonal therapy have achieved a significant
improvement in clinical outcomes and response rates.
In this manuscript we will chronologically review major milestones in
the evolution of endocrine
therapy over the last 100 years, highlighting each class of agent and
key trials that have led
to changes in modern practice.
Keywords: Breast cancer; estrogen receptor;
endocrine therapy
Introducción
El cáncer de mama (CM) es la neoplasia maligna
más común en mujeres. En el 2018, se diagnosticaron
2.088.849 casos y hubo 626.679 muertes asociadas a
esta enfermedad (1). Se estima que para el año 2020,
en el mundo habrá cerca de 2 millones de casos nuevos de CM, lo que
representa un incremento del 26%,
de los cuales 76% se presentarán en países de ingresos
medios y bajos, y de estos un 20% en América Latina
(2,3). En la actualidad el CM constituye un problema
en salud pública en Latinoamérica, ya que el número de casos y la
mortalidad han ido en aumento, particularmente en países del cono sur
(3). En México, la
proporción de mujeres con CM pasó de 14,17 a 25,17
por cada 100.000 mujeres entre el año 2001 y 2011
(4). En Colombia la incidencia también aumentó de
27,8 a 49,7 casos por 100.000 personas en el periodo
de 2007 a 2012 (5), representando el 13% de todas las
neoplasias con 13.380 casos nuevos y 3.702 muertes
asociadas (2).
El CM es una enfermedad principalmente de mujeres
postmenopáusicas, con una edad media de presentación de 62 años y menos
del 10% diagnosticado antes
de los 45 años (6). Sin embargo, en América Latina la
incidencia en mujeres menores de 44 años es mayor
comparada con países desarrollados (20% vs. 12%) (7).
La mayoría de los CM son carcinomas, siendo el carcinoma ductal
infiltrante el subtipo más común (50-75%),
seguido del carcinoma lobulillar infiltrante (5-15%) (8).
Cuando hablamos de CM hacemos referencia a una
enfermedad heterogénea, no solo en sus características
clínicas, sino también biológicas. Estudios de perfiles de
expresión génica han identificado al menos 5 subgrupos histomoleculares
diferentes (luminal A, luminal B,
subtipo-basal, Her2+ y subtipo normal like), cada uno
con un comportamiento clínico distinto y una respuesta variable al
tratamiento (9,10). Se ha propuesto una
clasificación molecular simplificada basada en el perfil
de inmunohistoquímica, utilizando la expresión de receptores hormonales
(RH), el estado del receptor del
factor de crecimiento epidérmico humano -2 (Her2) y
marcadores de proliferación, clasificando a los subtipos
en luminal A y B, triple negativo y Her2 (enriquecido
con Her2 y Her2/luminal) (11).
De acuerdo con los datos recientes del Instituto Nacional de Cáncer en
Estados Unidos, al momento del
diagnóstico el 60% de los casos son localizados, un
30% localmente avanzados y aproximadamente un
10% metastásicos, con una supervivencia global (SG)
a 5 años del 98%, 85,7% y 28% respectivamente (6).
El desarrollo de la terapia endocrina ha transformado el tratamiento de
los pacientes con CM. El cambio
de la cirugía ablativa y las quimioterapias agresivas
por una terapia dirigida y mejor tolerada ha mejorado tanto la
mortalidad como la calidad de vida de las
pacientes con CM hormonosensible. En los siguientes
párrafos, hacemos un relato de los hitos clave en la
evolución del tratamiento hormonal durante el último
siglo, destacando cada clase de agente y los ensayos
clave que han llevado a cambios en la práctica clínica
(
Tabla 1).
Tratamientos ablativos quirúrgicos
Los primeros tratamientos endocrinos para mujeres
con CM avanzado datan de más de un siglo y se trataban de tratamientos
ablativos quirúrgicos incluyendo:
ooforectomía bilateral, adrenalectomía bilateral e hipofisectomía
(12,13).
Albert Schinzinger, un cirujano de origen alemán, fue
el primero en proponer la ooforectomía bilateral como
tratamiento para el CM avanzado, esto bajo la observación de que las
mujeres jóvenes cursaban con una
enfermedad más agresiva, por lo que sugirió que estas
deberían hacerse “mayores” mediante la extirpación
de los ovarios (14,15). George Thomas Beatson (13)
fue la primera persona en realizar una ooforectomía
bilateral en una mujer con CM en 1895. Reportó 3
casos de mujeres con CM avanzado tratadas con ooforectomía bilateral
(13), de las cuales 2 presentaron
regresión posterior al manejo quirúrgico, describiendo
una posible asociación entre el ovario y el desarrollo
del CM, aunque no sabía la razón de su éxito. Desafortunadamente, esta
práctica no beneficiaba a todos los
pacientes y, en 1900, Stanley Boyd demostró que sólo
un tercio respondían a la ooforectomía bilateral, y que
las respuestas duraban en promedio de 1 a 2 años (16).
A pesar de estos resultados desalentadores, a principios del siglo XX
la ooforectomía bilateral se convirtió en un estándar de tratamiento,
aunque nunca fue una práctica popular por la alta morbilidad asociada
(15,16).
Tabla 1. Descripción de terapias endocrinas en cáncer de mama
avanzado.
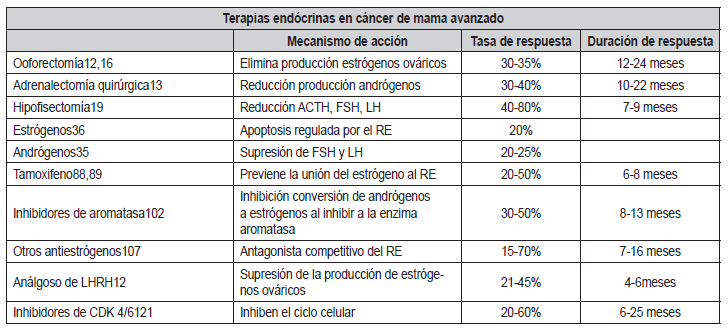
ACTH: hormona adrenocorticotrópica, FSH: hormona folículo
estimulante, LH: hormona luteinizante, LHRH: hormona liberadora de
hormona luteinizante, RE: receptor de estrógeno, CDK: ciclina
dependiente de cinasa.
A mediados de los años 1950’s, se introdujeron dos
nuevas estrategias de tratamiento, la adrenalectomía
bilateral y la hipofisectomía. Charles Huggins (17)
introdujo la adrenalectomía bilateral seguido de sustitución con
cortisol como una nueva estrategia de tratamiento. Se propuso que
existía un componente hormonal que se originaba en las glándulas
suprarrenales
y que sostenía y propagaba la enfermedad en ciertos
tipos de CM. Posteriormente, numerosas investigaciones confirmaron el
efecto benéfico de la adrenalectomía en la regresión tumoral (18) tasas
de respuestas de
entre 30-40% y SG entre 10-22 meses (13).
En 1957, Luft y Olivecrona (19) reportaron que más del
50% de sus pacientes con CM avanzado se beneficiaban de realizarles
hipofisectomía, y señalaron además
que las pacientes con enfermedad indolente y aquellas
que habían respondido al tratamiento endocrino previo
tenían mejores resultados después de la hipofisectomía
(tasa de respuestas: 50-80% y SG 6 meses – años), esto
a expensas de una alta tasa de mortalidad (5-10%) (20,
21). Sin embargo, para entonces no había medios para
predecir qué pacientes con CM responderían. No fue
sino hasta 1962, cuando Elwood Jensen
et
al (16) descubrieron la expresión de receptores de estrógeno en
el
tejido mamario como predictor de la respuesta a la terapia hormonal
(16,22). Debido a la alta morbilidad, los
tratamientos ablativos originales se reservaron para su
uso en pacientes que experimentaban una recaída después de la supresión
ovárica primaria (12).
Mas adelante, en los años ochentas, los agonistas de
la hormona liberadora de hormona luteinizante, que
reducían la producción de estrógenos ováricos y sin
la necesidad de realizar una cirugía fueron aprobados
como tratamiento para mujeres premenopaúsicas en
mujeres con CM avanzado (12).
Identificación y cristalización de
estrógenos (síntesis y evaluación en el
laboratorio)
En 1929, el fisiólogo reproductivo Edgar Allen, quien
estudiaba los cambios cíclicos ocurridos en el útero de
ratones con ooforectomía bilateral (23), describió que
el desarrollo del folículo ovárico precedía la aparición
del estrato córneo en la vagina y que, inyectando fluido folicular,
dichas células escamosas volvían a identificarse (24), demostrando el
sitio de origen de la hormona. Con ayuda del bioquímico orgánico Edward
Doisy (25), lograron purificar por primera vez la estrona utilizando
una mezcla de soluciones acuosas ácidas
y el extracto de folículo ovárico. Dicho extracto, una
vez combinado con éter etílico y luego de varios procesos, pudo ser
transformado en cristales que fueron
purificados posteriormente. Estos resultados fueron
presentados de forma preliminar en el 13° Congreso
Internacional de Fisiología de Boston (26) y formalmente en un artículo
en 1930 (27).
Al mismo tiempo y de forma independiente, Adolph
Butenandt lograba la misma hazaña (28); pero fue a
este último a quien le fue otorgado el Premio Nobel
(junto con Leopold Ruzincka) por este descubrimiento, ya que fue el
primero en publicar sus resultados en
agosto de 1929 (29).
Estrógenos y su relación con el cáncer
El descubrimiento de los estrógenos (estriol, estrona
y estradiol) a partir de orina de mujeres embarazadas
por Butenandt y Doisy (16), proporcionó el ímpetu
para comprender mejor la estructura, biosíntesis, secreción y función
de los diversos estrógenos.
Durante las décadas siguientes, se acumularon observaciones de que una
pequeña cantidad de estas hormonas podría causar un crecimiento
profundo de diversos
tejido diana. Los estrógenos están involucrados en una
gran cantidad de procesos fisiológicos del cuerpo, así
como también están relacionados con algunas patologías (30) MER-25
(ethamoxytriphetol. Por lo tanto,
está claro que la esteroidogénesis ovárica tiene gran
importancia en el desarrollo normal de la mama y en
la génesis del CM (31).
Como hemos visto, el vínculo entre los estrógenos y
el crecimiento y desarrollo del CM se ha reconocido
durante más de un siglo, y la dependencia del CM en
la acción estrogénica se reconoció por primera vez mediante la
regresión inducida por la ooforectomía bilateral en mujeres
premenopáusicas (13). Posteriormente,
se correlacionó la función ovárica con la producción
de estrógenos y el aislamiento de la proteína receptora
de estrógenos, combinado con una mayor incidencia
de tumores con receptores de estrógeno (RE) positivos
en mujeres posmenopáusicas. A su vez, esto condujo a
la identificación de una fuerte asociación entre la dosis
de estrógeno y la duración de la exposición a este con
un mayor riesgo de CM (31). Es por lo tanto lógico que
el siguiente paso histórico haya sido la identificación
de fármacos que actuaran sobre los estrógenos y sus
receptores como tratamiento del CM.
Tratamientos ablativos hormonales
Probablemente uno de los éxitos más importantes
en el tratamiento del CM avanzado a principios del
siglo XX fue la alteración del entorno endocrino interno tumoral a
través de la terapia hormonal. El
fundamento de este tratamiento es la dependencia
bien demostrada de la mama a la estimulación hormonal (21).
Los primeros intentos de alterar el curso del CM en el
ser humano mediante la administración de hormonas
sexuales fueron ambiguos (32). En 1935, Haddow
et
al. observaron que algunos hidrocarburos cancerígenos con una
ligera actividad estrogénica retardaban el
crecimiento de los carcinomas mamarios, por lo que iniciaron ensayos
terapéuticos con los propios estrógenos en el tratamiento del CM
avanzado (21,32).
El primer informe clínico del uso de andrógenos en
el CM metastásico fue el de Ulrich y Loeser en 1939,
que describieron el uso de propionato de testosterona
para tratar el CM avanzado (33). La administración
de esteroides androgénicamente activos a mujeres con
CM avanzado se volvió una práctica común en ese entonces: se
administraban 100 mg intramusculares de
manera trisemanal, con lo que se lograban tasas de
respuesta del 20-25%, con una mejoría en el control de
síntomas del 50% (21,34,35). Los andrógenos podían
utilizarse tanto en el período premenopaúsico como
en el posmenopáusico; y los estrógenos solo en pacientes
posmenopáusicas.
A mediados de los años 1940’s, y de manera paradójica, los estrógenos
también formaban parte del arsenal
del tratamiento del CM, principalmente para aquellas
pacientes con lesiones en tejidos blandos (12).
Los primeros estudios realizados fueron con estrógenos sintéticos no
esteroideos en dosis altas, como el
etinilestradiol y dietilestilbestrol (DES)16 con lo que
se lograba regresión tumoral hasta en un 20% de los
casos, aunque a costa de una alta toxicidad (36).
Desafortunadamente, las pacientes que tenían una
respuesta inicial favorable, eventualmente tenían un
recrudecimiento del proceso de la enfermedad en meses o años (32). En
la actualidad ambas terapias están
proscritas.
Desarrollo temprano de
antiestrógenos y eficacia en el
tratamiento del CM
El descubrimiento de hormonas estrogénicas producidas en el ovario por
Edgar Allen y Edward Doisy en
1923 (37) impulsó la búsqueda de un antagonista terapéutico para
reducir la incidencia de CM en individuos
predispuestos a la enfermedad por su sensibilidad a las
hormonas estrogénicas. Los primeros antiestrógenos
no esteroideos MER-25 y clomifeno, fueron sintetizados como reguladores
de la fertilidad en pacientes con
anovulación, y posteriormente se descubrió su utilidad
en otras patologías como el CM (16,30).
En 1958, Leonard Lerner
et al (38)
publicaron un artículo de referencia sobre las propiedades
farmacológicas del primer antiestrógeno no esteroideo, el
etamoxitrifetol (MER-25). Este compuesto fue descubierto de
forma serendípica al encontrarse que tenía una estructura similar a la
de los estrógenos (trifeniletileno), y
que tenía una acción antiestrogénica débil pero constante en todos los
modelos animales (39). Los doctores
Roy Hertz de los Institutos Nacionales de Salud de los
Estados Unidos (NIH) y Robert Kintner de la Universidad de Harvard,
fueron los primeros en investigar su
utilidad en el tratamiento de CM con resultados alentadores. Sin
embargo, los estudios clínicos posteriores
demostraron que el fármaco era demasiado tóxico para
el uso general (causando alucinaciones, episodios psicóticos,
pesadillas y alteraciones del sueño entre otros)
(30). Posteriormente, se descubrió que un compuesto
sucesor, el clomifeno (una mezcla de isómeros geométricos estrogénicos
y antiestrogénicos de un trifeniletileno) también era inductor de la
ovulación en mujeres
subfértiles (38). En 1964, Herbst y colaboradores (40)
demostraron que el citrato de clomifeno podría tener
eficacia en pacientes con CM. Sin embargo, la evaluación clínica de
estos compuestos para esas indicaciones nunca se llevó a cabo debido a
la concentración
del esfuerzo en el uso de clomifeno en la inducción de
la ovulación en mujeres infértiles anovulatorias (30).
En 1960, se patentó trifeniletileno como una terapia
anti-CM, siendo la nafoxidina, uno de sus derivados,
la base estructural del lasofoxifeno, el primer modulador selectivo del
RE. Posteriormente, se descubriría el
tamoxifeno como base fundamental de la terapia hormonal en mujeres con
CM (39).
Descubrimiento del receptor de
estrógeno – métodos de identificación
y tipos de ensayo
A partir de la observación de que la cantidad de estrógeno requerida
para su actividad fisiológica era mínima,
Edward Jensen intuyó que probablemente influenciaba
la reacción de numerosas moléculas que participaban
en los fenómenos metabólicos (41) y observó la naturaleza temporal de
la respuesta estrogénica, y la ausencia
de oxidación de estradiol a estrona en vivo, es decir, el
ejercicio de su actividad sin transformación metabólica
(42). En 1960, junto con Jacobson, utilizando estradiol
marcado con radio-isótopos, demostraron la absorción
específica del mismo en el útero y la vagina de ratones
castrados (22,43,44). Trabajos subsecuentes por Noteboom y Gorski
demostraron que el sitio de unión era
estereoespecífico y que podría tratarse de una proteína
con actividad de nucleotidasa, de unión reversible (45)
y más adelante, Toft y Gorski aislarían una macromolécula con las
características esperadas en un receptor
(46). En 1980, Jensen y Green produjeron anticuerpos
monoclonales para determinar la existencia de proteínas receptoras de
unión a estrógeno intracelulares (47) y
más adelante, King utilizó dichos anticuerpos en tejido
congelado de humanos donde observó tinción específica nuclear (48).
En 1986 el grupo liderado por Chambon logró clonar
el gen del RE (49) y, utilizando estudios de mutagénesis, encontraron
que el receptor consta de dominios de
unión a DNA que contienen motivos dedos de zinc y
dominios de unión a ligando (50), elementos estructurales clave de los
factores de transcripción dependientes de ligando (51).
En 1966, un segundo RE fue identificado por Gustafsson y colaboradores.
Este RE se expresaba predominantemente en células epiteliales de
testículo y
ovario y era homólogo al RE previamente descubierto
en ciertas regiones (52). El primero fue renombrado
como REα y este último fue denominado Reβ (
Figura
1) (51).
En CM, el grupo de Jensen determinó que el consumo
de estradiol era mayor que el detectado en el tejido mamario normal y
que era posible determinar la expresión
de RE en el tejido neoplásico, la cual es de predominio
nuclear. Con ello definieron dos subtipos de tumores
mamarios: RE positivos y RE negativos (53). Posteriormente, junto con
otros grupos, observaron que medir la cantidad de receptor en el tumor
(evaluado en
la mastectomía), podía ayudar a predecir la respuesta
a tratamientos hormonales y la recurrencia (54–58),
para lo cual propusieron dos tipos de inmunoensayos
(22): 1) Enzimático: mide la cantidad de receptor en el
citosol, cuantificada a través de colorimetría usando
una curva estandarizada; 2) Inmunocitoquímico: Usa
cortes de micras de espesor del tumor, a los que se les
aplica el método de peroxidasa-antiperoxidasa, con el
cual se tiñe el núcleo de las células donde se localiza
el receptor. Este último es el más utilizado en la actualidad.
Con lo anterior, fue posible identificar mejor a las pacientes que se
beneficiarían de terapia hormonal y a
su vez, disminuir tratamientos innecesarios a aquellas
que no obtendrían ningún beneficio (59, 60).
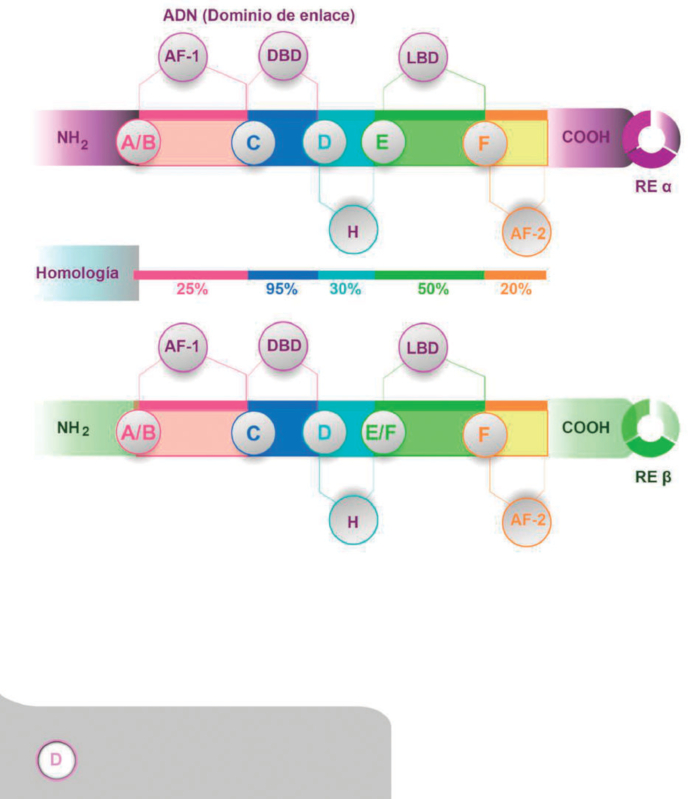
Figura 1. Se muestran los seis dominios funcionales de ambos
receptores de estrógeno (RE); así como la
homología entre ambos. AF-1 y AF2: Dominios de activación funcional
transcripcional 1 y 2. DBD: Dominio de unión
a ADN. H: Dominio de bisagra. LBD: Dominio de unión a ligando 61.
Mecanismo de acción de los
estrógenos
Ambos receptores de estrógeno (REα y REβ), pertenecen a la superfamilia
de receptores nucleares. Contienen seis dominios funcionales: amino
terminal (A/B),
de unión a ADN (C), de bisagra (D), de unión a ligando (E) y
carboxi-terminal (F); ambos receptores son
homólogos en los dominios C y E pero divergen en
A/B, D y F (Figura 1). REα se expresa mayormente
en útero e hipófisis y en menor cantidad en hígado,
hipotálamo, hueso, glándula mamaria, cérvix y vagina
(29). A pesar de que la descripción del descubrimiento de todas las
vías de señalización del RE está fuera del
ámbito de este artículo, un esquema detallado de los
principales mecanismos de acción del RE se muestra
en la
Figura 2.
Aunque ambos receptores pueden encontrarse en el tejido mamario normal,
REα sólo se encuentra en el núcleo de algunas células epiteliales y
regula el crecimiento y la elongación ductal (62); además, se ha
encontrado
en 50-80% de los tumores mamarios y su expresión se
correlaciona con factores de buen pronóstico (63);
mientras que REβ se localiza en células epiteliales y estromales de la
glándula mamaria normal y su expresión
se ha visto disminuida en algunas neoplasias invasoras
de esta región; además, algunos estudios sugieren que
inhibe la función transactivadora de REα, lo que le confiere
propiedades antiproliferativas y otros han asociado
la expresión positiva del receptor en células neoplásicas
con un pronóstico favorable; sin embargo, aún hace falta más
investigación en esta área (64).
Desarrollo de modelo murino
para la investigación de CM
hormonodependiente
Gracias al trabajo de muchos investigadores que sentaron las bases
(74-79), en 1958 Huggins presentó el
primer modelo murino en el que era posible producir
carcinomas mamarios en un período menor a 30 días
al administrar 3-metilcolantrano (un compuesto aromático) a través del
tracto gastrointestinal de ratas.
Con ello logró demostrar tres hitos en el estudio del
CM: 1) la mayoría de los tumores producidos por este
método eran hormono-dependientes (cuando se retiraba el soporte
hormonal interno del animal, la mayoría
de los tumores disminuían de tamaño, y esta disminución era debido a la
atrofia de las células epiteliales y
no a necrosis de las mismas; 2) la administración de
dihidrotestosterona también producía atrofia en las células
neoplásicas, mientras que inducía proliferación
del tejido mamario no neoplásico; 3) existía un porcentaje bajo de
tumores que continuaban creciendo a
pesar del retiro de soporte hormonal, pero aún en estos
se podían identificar áreas de atrofia coexistiendo con
zonas de proliferación, por lo que no todas las células
en un mismo tumor tenían la misma respuesta al tratamiento (80).
Estos hallazgos fueron de suma relevancia, ya que
aunque el concepto de dependencia hormonal había
sido establecido previamente en tumores provenientes
de humanos, no había sido conseguido en modelos
murinos; además, gracias a estos estudios fue que nació el concepto de
terapia hormonal (81).
Desarrollo del tamoxifeno y actividad
antitumoral
En los años setentas el tamoxifeno fue el primer antiestrógeno no
esteroideo que representó un cambio radical en el tratamiento del CM.
El compuesto ICI 46474
(tamoxifeno) se sintetizó por primera vez en 1963
como un medicamento anticonceptivo (82). Sin embargo, por su efectos
estrogénicos paradójicos también
fue estudiado como un medicamento para tratar la infertilidad por sus
efectos inductores de ovulación en
mujeres con amenorrea secundaria (83). Sin embargo,
serendípicamente se descubrió que tenía un efecto antiestrogénico, ya
que en estudios en ratas se encontró
que la forma transisomérica del compuesto prevenía la
implantación de embriones en el útero y causaba cornificación del
epitelio vaginal como un efecto atípico
al inhibir la respuesta de los estrógenos exógenos (82).
Estudios de laboratorio demostraron que, a nivel celular, el tamoxifeno
se unía al RE citoplasmático pero
que esto no provocaba síntesis de ADN a nivel nuclear
actuando como un falso mensajero al bloquear la actividad del RE (84).
En ratas se encontró que dependiendo de la dosis y del tejido en el que
actuaba podía
tener efectos pro o antiestrogénicos: en el tejido mamario tenía un
fuerte efecto antiestrogénico, mientras que en el epitelio uterino su
efecto era proestrogénico
(85). Más tarde, se confirmó que este efecto uterino aumentaba el
riesgo de cáncer de endometrio en mujeres
tratadas crónicamente con tamoxifeno (86).
Los efectos antitumorales del tamoxifeno fueron descritos por V. Craig
Jordan y colaboradores, al observar
que prevenía la inducción y crecimiento de tumores
mamarios inducidos por carcinógenos en ratas (87).
En 1969 se realizó el primer estudio con tamoxifeno en
pacientes con CM recurrente, encontrando remisión
hasta en el 22% de los casos (88). Posteriormente, se
llevaron a cabo otros estudios que confirmaron su efectividad en
pacientes con CM avanzado, utilizándose
principalmente en el contexto paliativo (89). Debido
a su baja incidencia de efectos adversos, el estudio del
tamoxifeno se extendió a etapas tempranas del CM,
y con más de 30 mil pacientes incluidas en estudios
aleatorizados se demostró una reducción de aproximadamente 25% en
recaída y 17% en mortalidad en tumores con receptores hormonales
positivos (90). Más
adelante, al observarse en los estudios en adyuvancia
que el uso de tamoxifeno disminuía la aparición de
tumores contralaterales (aunado a la prevención de
carcinognénesis en estudios con ratones), se llevaron
a cabo estudios utilizando tamoxifeno en mujeres sin
CM pero con alto riesgo de padecerlo, demostrándose
una disminución en la incidencia de CM, por lo que
se convirtió en el primer tratamiento preventivo para
cualquier tipo de cáncer, sentando los principios de la
quimioprevención (91,92).
Desde su aprobación por la Administración de Alimentos y Medicamentos
de los Estados Unidos (FDA)
en 1977, el tamoxifeno se ha utilizado para tratar a
millones de mujeres y hombres con CM RH positivos,
y sigue siendo tratamiento de elección en algunos escenarios. A lo
largo del tiempo ha demostrado tener efectos sobre la reducción de
riesgo de aproximadamente
50% para recaída o aparición de nuevos tumores en
pacientes con cáncer localizado (93). También se utiliza como
tratamiento neoadyuvante antes de la cirugía o tratamiento primario en
mujeres no candidatas
a cirugía. En el contexto metastásico ha demostrado
tener un efecto en el crecimiento de la enfermedad, y
adicionalmente, ha demostrado capacidad para disminuir el riesgo de
cáncer en mujeres con alto riesgo para
desarrollar cáncer.
Descubrimiento de la aromatasa
y producción extraglandular de
estrógenos
Desde los años treintas se tenía evidencia de que la
testosterona podía convertirse en “hormonas femeninas”, y el estudio de
la síntesis de los estrógenos permitió identificar metabolitos
precursores que a su vez llevaron al reconocimiento del proceso de
aromatización
(94). Sin embargo, fue hasta los años ochenta cuando
se logró la purificación de la enzima aromatasa, miembro de la familia
citocromo P450, por Yoshio Owasa
y colaboradores, y hasta el 2009 la cristalización de su
estructura por Ghosh y colaboradores (95).
Por otro lado, Paul MacDonald, Pentti Siiteri y colaboradores fueron
los primeros en describir la actividad
aromatasa extraglandular (94), demostrando que el tejido adiposo era
rico en aromatasa y la fuente principal
de síntesis de estrógenos en mujeres posmenopaúsicas
(96). Además, estudios en mujeres posmenopáusicas
mostraron que la conversión de androstendiona a estrógeno era mayor en
sujetos con obesidad (97), lo que
más adelante fue de interés para investigadores como
un blanco terapéutico tanto para el CM como para
otras patologías como la infertilidad.
Desarrollo de inhibidores de
aromatasa esteroideos y no
esteroideos
En los años setentas, Harry y Angela Brodie probaron
más de 100 inhibidores de aromatasa (IA) esteroideos, lo que llevó a la
identificación de la 1,4,6-androstatriendiona y la 4-hidroxi
androstenediona (4-OH-A)
como los candidatos más prominentes (98). En colaboración con Charles
Coombes, Paul Goss y Mitch
Dowsett se lanzó el primer ensayo clínico con un IA
selectivo (4-OH-A o formestano) para el tratamiento
del CM en el Hospital Royal Marsden en Londres, Inglaterra,
demostrándose su eficacia en pacientes que
habían progresado al tratamiento con tamoxifeno (99).
Más adelante, se reconoció que el formestano no era lo
suficientemente potente para bloquear a la aromatasa,
y se buscaron inhibidores más potentes.
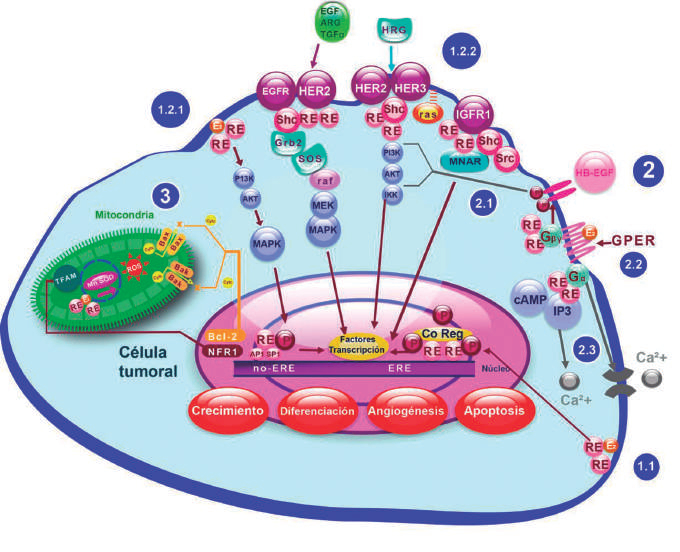
Figura 2. Resumen de las vías de señalización del receptor de
estrógeno (RE) descubiertas hasta el momento. 1)
Vía genómica (nuclear): 1.1) Clásica: En esta vía el estrógeno se une
al receptor para promover su dimerización
y fosforilación (65), lo que permite la unión del complejo “ligando-RE”
con co-reguladores esteroideos (Co Reg) y
elementos de respuesta a estrógenos (ERE). 1.2) Vía alterna: 1.2.1)
Independiente de ERE: El complejo “ligando-RE”
interactúa con otros factores de transcripción como la Proteína
Activadora 1 (AP1) y la Proteína Específica 1 (SP1) (66).
1.2.2) Independiente de ligando: Mediada por factores de crecimiento
(EGFR, HER2, y receptor de factor de crecimiento
similar a la insulina tipo 1 (IGFR1)), que activan cascadas de
proteínas cinasas (Ras-ERK; PI3K-Akt)66. Todas finalizan
con cambios en la transcripción génica que regulan el crecimiento,
diferenciación, apoptosis y angiogénesis celular;
acciones moduladas por la estructura del ligando, el subtipo de
receptor e isoforma, el gen promotor y el balance de
co-activadores y co-represores (67,68). 2)Vía no genómica (citosólica)
(69): Comienza con la union del ligando al
Receptor de Estrógeno Unido a Proteina G (GPER) que lleva a la
liberación de las subunidades α y βγ de proteínas G
heterotriméricas. 2.1) Activación de cascadas de proteínas cinasas: La
subunidad βγ activa metaloproteasas de matriz
que liberan el EGF unido a Heparina, el cual es autofosforilado al
unirse a su receptor, que activa las MAP cinasas
Erk1/2 (induce transcripción de c-fos, Egr-1, ERRα y aromatase en el
núcleo) y PI3K (fosforila la cinasa Akt que fosforila
a su vez al factor de transcripción FOXO3a en el núcleo). 2.2)
Activación de ciclina de Adenosin Monofosfato(cAMP):
La subunidad α activa Adenolil Ciclasa en el citosol, que genera cAMP,
el cual activa la Proteína Cinasa A que fosfolila
a la proteína elemento de respuesta de unión con cAMP (CREB) que
modifica la expresión de ciertas proteínas (Ej.
Ciclina D1, que promueve el progreso del ciclo celular). 2.3)
Activación de vías de calcio: La liberación de la subunidad
α de proteínas G heterotriméricas activa la Fosfolipasa C, que une
Fosfatidilinositol 4,5-bifosfato a diacilglicerol e IP3.
Este último libera calcio, el cual activa diversas enzimas en el
citosol y activa Erk1/2. La unión de estrógenos a GPER
también facilita la apertura de canales L de calcio en la membrana
citoplásmica. 3) Interacción núcleo-mitocondria:
La transcripción nuclear de NRF1(Factor Respiratorio Nuclear 1)
promueve la transcripción del factor mitocondrial
TFAM (Factor FA de Mantenimiento del ADN mitocondrial) que a su vez
regula genes codificados en el mtADN (70).
En algunas células, RE interactúa con el loop-D del mtADN, lo que
incrementa directamente la actividad de la enzima
recolectora de ROS MnSOD (Manganasa-superóxido dismutasa) y del gen
GPX1 (glutatión peroxidasa) que las
protegen de apoptosis inducida por oxidación mediada por ROS (especies
reactivas de oxígeno) (71). La interacción
también es indirecta, a través de las proteínas de la familia de Bcl2,
que controlan la integridad de la membrana externa
mitocondrial impidiendo la salida de Citocromo-C (clave para la
apoptosis) (72). Los estrógenos protegen a la célula de
la apoptosis al favorecer la transcripción de Bcl2 (73).
Con el reconocimiento de la inhibición de la aromatasa como un blanco
importante para el tratamiento del
CM, varias compañías farmacéuticas contribuyeron
con la identificación y el desarrollo clínico de inhibidores
esteroideos y no esteroideos de la enzima aromatasa. El primer agente
de segunda generación fue
el inhibidor CGS-16949A, fadrozole, que inhibía más
eficazmente la producción de estrógenos pero inesperadamente tenía
también efecto al bloquear a la aldosterona y nunca tuvo un uso clínico
(100,101). Posteriormente, gracias a estudios preclínicos en ratones y
cultivos celulares de CM (MCF.7Ca), se desarrollaron
dos inhibidores de tercera generación reversibles no esteroideos, el
anastrazol y el letrozol, y un inhibidor esteroideo, el exemestano.
Diversos estudios mostraron
que estos IA eran más efectivos y con un efecto más
prolongado para reducir el volumen de los tumores
que el tamoxifeno (94).
Comparación entre los antiestrógenos
y los IA
Estudios colaborativos, multicéntricos, multinacionales y aleatorizados
que incluyeron alrededor de diez
mil pacientes con CM, compararon directamente
la efectividad de los IA contra el tamoxifeno en pacientes con cáncer
avanzado (102). Todos demostraron superioridad en eficacia clínica, con
un perfil de
toxicidad distinto. Los IA se asociaron con un incremento en el riesgo
de osteoporosis, osteopenia, artralgias y mialgias, mientras que el
tamoxifeno se asoció
con un mayor riesgo de trombosis venosa profunda y
tromboembolia pulmonar. Estos estudios colocaron a
los IA como primera línea en el tratamiento del cáncer avanzado.
Posteriormente, iniciaron los estudios
en el contexto de enfermedad temprana con distintos
diseños: comparación directa con tamoxifeno (ATAC,
BIG-FEMTA),
switch (MA-17,
ABCSG-ARNO, BIG1-98) y en la neoadyuvancia. En todos ellos también
se demostró superioridad de los IA, y actualmente se
consideran el tratamiento de primera elección para la
adyuvancia en mujeres posmenopaúsicas (103). Recientemente, también se
ha probado su eficacia en estudios
de prevención para pacientes con alto riesgo de desarrollar cáncer
(104).
Antagonistas del receptor de
estrógeno (fulvestrant)
El desarrollo de moléculas pequeñas para el tratamiento del cáncer se
ha enfocado en encontrar compuestos que ocupen sitios de unión que
afectan de
forma directa la unión de proteínas. Sin embargo,
otra opción es la inducción de degradación de proteínas mediante la
desestabilización de receptores (105).
Dado que los moduladores de los receptores de estrógenos pueden tener
efectos diversos dependiendo
de su sitio de acción, este tipo de fármacos parece
particularmente interesante para lograr la inhibición
selectiva de estos receptores causando menos efectos
adversos.
El descubrimiento de que la adición de un grupo decametileno en la
posición 7α del estradiol podía generar
un antiestrógeno puro sin inhibir la unión con el receptor, llevó a la
búsqueda de compuestos que carecieran
de efectos sobre el endometrio, encontrándose así el
ICI182780, ahora conocido como fulvestrant (106).
Estudios
in vivo e in vitro
demostraron que el fulvestrant tenía una potencia antiestrogénica
incluso superior al
tamoxifeno, y posteriormente los ensayos clínicos han
demostrado que, en pacientes con CM avanzado, este
compuesto tiene mayor tasa de beneficio clínico que
otras hormonoterapias (107).
Cinasas dependientes de ciclinas y
desarrollo del CM
La pérdida de control del ciclo celular es una de las
principales características de las células malignas. En
el CM en específico, el estudio del ciclo celular y de
los diferentes puntos de control y vías de regulación
ha llevado a entender algunos mecanismos de resistencia a la
hormonoterapia. A principios de los años
noventa, estudios realizados en líneas celulares de CM
detectaron la sobreexpresión de diversas ciclinas, particularmente D1 y
E1, así como una asociación entre
esta sobreexpresión y los desenlaces negativos de los
pacientes (108).
Estudios adicionales detectaron que hasta el 45% de las
biopsias tumorales de mama expresaban altos niveles
de mARN de Ciclina D1, por lo que se consideró que
esta unidad regulatoria del ciclo celular participaba de
forma predominante en el control de la proliferación
celular en CM (109). Así mismo, se observó que uno
de los factores más importantes para la oncogénesis
en CM era la fosforilación de las cinasas dependientes
de ciclinas (CDK) CDK4 y CDK6, que a su vez tenía
como consecuencia la activación de la proteína del retinoblastoma (Rb)
(110). Este fenómeno, acoplado a la
desregulación de la expresión de ciclina D1, conducía
a la progresión del ciclo celular y a la proliferación oncogénica
(111).
Inhibidores de cinasas dependientes de
Ciclina CDK 4/6
Los primeros intentos farmacológicos se centraron en
generar inhibidores no específicos de CDK, también
conocidos como inhibidores “pan-CDK”. El primero,
y más estudiado, de dichos inhibidores fue el flavopiridol, que se
utilizó en más de sesenta estudios en diferentes tipos de tumores
(112). Sin embargo, los intentos iniciales fueron desalentadores, ya
que las tasas de
respuesta in vivo no se correspondieron con el efecto in
vitro. La segunda generación de inhibidores se caracterizó por ser más
específica, intentándose bloquear de
forma selectiva algunas CDK, principalmente CDK1
y CDK2 (110). Dinaciclib, un inhibidor selectivo de
CDK1, CDK2, CDK5 y CDK9, mostró beneficio en
algunos tumores, pero tuvo resultados negativos en un
ensayo aleatorizado fase II en CM (113). Los estudios
iniciales de inhibidores de CDK, por lo tanto, no cumplieron con sus
objetivos, principalmente debido a que
su potencial de inhibición era leve y a la inadecuada
selección de pacientes, ya que se estudiaron muchos
tipos de tumores al mismo tiempo.
Para la tercera generación de inhibidores de CDK, el
interés se centró en el desarrollo de fármacos que inhibieran CDK4 y
CDK6, con la intención de generar la detención citostática del ciclo
celular en G0/G1. Esto llevó
al descubrimiento por parte de la empresa Pfizer del compuesto PD
0332991, que mostró una inhibición altamente específica de CDK4 y CDK6
después de su administración oral en ratones, y que hoy en día se
conoce como
palbociclib (114,115). Al mismo tiempo, Novartis llevó a
cabo la demostración de la utilidad del inhibidor LEE011
(después conocido como abemaciclib) en líneas celulares
de neuroblastoma116 y liposarcoma (117), mientras que
Eli Lilly mostró que otro inhibidor de CDK4 y CDK6,
LY2835219 (ribociclib) tenía actividad farmacológica sobre células de
melanoma, tanto solo como en combinación con quimioterapia (118, 119).
El siguiente paso fue la utilización de estos fármacos
en el CM. Esto se vio motivado por la observación de
que la resistencia al tratamiento endócrino parecía deberse a la falta
de regulación en genes asociados a la
proliferación que se encuentran regulados por el eje CDK4/6-Rb (120).
Hasta el día de hoy, los tres inhibidores de CDK4 y CDK6 han sido
utilizados en por
lo menos ocho ensayos clínicos aleatorizados fase III y
en un estudio aleatorizado fase II en CM metastásico
con RH positivos. En conjunto, estos estudios demuestran que,
comparados con terapia hormonal en monodroga, el tratamiento con
inhibidores de CDK4/6 más
terapia hormonal se asocia con una mejoría significativa en SG,
supervivencia libre de progresión y tasa de
respuesta en pacientes con CM RH positivos (121).
Identificación de mutaciones en RE
en pacientes con CM tratadas con
hormonoterapia
Con el advenimiento de las terapias hormonales, comenzaron también
múltiples estudios para evaluar
su efectividad. En retrospectiva se observó que era
posible alcanzar regresión tumoral en más del 80%
de las pacientes, pero estas duraban 12-18 meses
en promedio y posteriormente, usualmente ocurría
adaptación tumoral y recaída (122). Santen y colaboradores propusieron
que lo anterior podría deberse a
hipersensibilidad al estradiol por activación de MAP
cinasa (123).
En 2009, Fuqua y colaboradores identificaron una
transición de lisina por arginina en el residuo 33 del
REα (K303R) en lesiones malignas y premalignas de
la mama que les confería hipersensibilidad a estrógenos y resistencia a
tamoxifeno, y propusieron que el
mecanismo de resistencia involucraba una unión entre
el receptor mutante y la subunidad reguladora p85α
de la cinasa fosfatidilinositol-3-OH (PI3K) que llevaba al incremento
de su actividad y activación de la
vías de supervivencia de PKB/Akt; proponiendo esta
mutación como un marcador predictor de respuesta a
tratamiento, y al bloqueo de la vía PI3K/Akt como
una estrategia de tratamiento en neoplasias resistentes
a terapia hormonal (124).
A partir de ese momento, diversos investigadores identificaron
mutaciones en la subunidad α del RE (125,126)
o en su ligando (127,128) hasta en 20% de las neoplasias
que progresan después de terapia hormonal (129); proponiéndose como
mecanismos clave en la activación
del receptor independiente de estrógenos y la resistencia
endocrina adquirida (130).
Presente y futuro del tratamiento del
CM
En los países desarrollados del mundo, donde el acceso al tratamiento
del CM está menos obstaculizado
por barreras del sistema de salud, la mortalidad por
CM ha disminuido progresivamente entre los años noventas del siglo
pasado y la actualidad (9,131). En el
CM metastásico con RH positivos, específicamente, la
supervivencia prácticamente se ha duplicado, pasando
de 32 a 57 meses (132). Aunque esta mejoría puede ser
atribuible a muchos factores, no hay duda de que el
desarrollo de nuevos fármacos ha tenido un papel fundamental. Sin
embargo, todavía existen importantes
preguntas que contestar, particularmente en aquellas
pacientes que desarrollan resistencia al tratamiento
hormonal. Las vías futuras de investigación incluyen
el estudio y tratamiento de tumores con mutaciones
en el RE, el hallazgo de biomarcadores, y el uso de
biopsias líquidas, con la intención de desarrollar tratamientos
personalizados que permitan mejorar la supervivencia de estas
pacientes.
Conclusiones
La introducción y posterior evolución de muchos
agentes endocrinos desde la década de 1970 ha transformado el
tratamiento de las mujeres con CM (
Figura
3). Estos avances han logrado un cambio en la
práctica clínica actual, pasando de cirugías ablativas
y quimioterapias agresivas, a tratamientos dirigidos y
mejor tolerados, lo que ha mejorado la supervivencia y
la calidad de vida de las pacientes con CM.
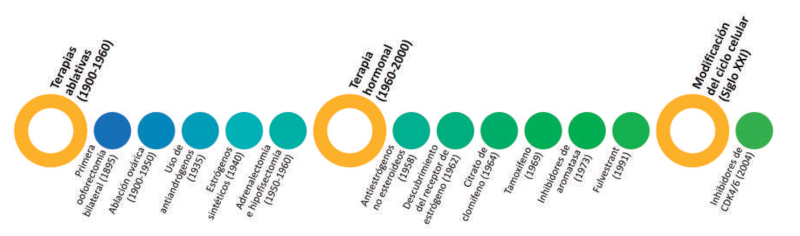
Figura 3. Evolución cronológica de los descubrimientos más
importantes en el tratamiento
del cáncer de mama con receptores hormonales positivos.
Conflictos de interés
Yanin Chávarri Guerra ha recibido financiamiento
para investigación de Roche y apoyo de viáticos de
Pfizer. Los demás autores se declaran sin conflictos de
interés.
Agradecimientos
Agradecemos a la diseñadora Luí Balmonte por el diseño y elaboración de
las figuras 1 y 2.
Referencias
1. (IARC) IA for R on C, World Health
Organization (WHO).
Breast Cancer. Source: Globocan 2018. World Heal Organ.
2018;876:2018-2019.
2. Bloom D. Breakaway: The global
burden of cancer -challenges and opportunities. A report from the
Economist
Intelligence Unit Limited 2009. London: The Economist;
2009.
3. Bravo LE, García LS, Carrascal E,
Rubiano J. Burden of
breast cancer in Cali, Colombia: 1962-2012. Salud Publica Mex.
2014;56(5):448-456.
4. Soto-Perez-de-Celis E,
Chavarri-Guerra Y. National and
regional breast cancer incidence and mortality trends in
Mexico 2001-2011: Analysis of a population-based database. Cancer
Epidemiol 2016;41:24-33
5. Duarte C, Salazar A,
Strasser-Weippl K, de Vries E,
Wiesner C Krush L et al. Abstract P5-13-12: Breast cancer in Colombia:
A growing challenge for the health care
system. Cancer Res.:1-6.
6. SEER*Explorer: An interactive
website for SEER cancer
statistics [Internet]. Surveillance Research Program NCI
[consultado 14 septiembre 2020].Disponible en:
https://seer.cancer.gov/explorer/.
7. Villarreal-Garza C, Aguila C,
Magallanes-Hoyos MC et
al. Breast Cancer in Young Women in Latin America: An
Unmet, Growing Burden. Oncologist. 2013;18(S2):26-34.
8. Waks AG, Winer EP. Breast Cancer
Treatment: A Review. JAMA. 2019;321(3):288-300.
9. Shumway DA, Sabolch A, Jagsi R.
Breast Cancer. Med
Radiol. 2020:1-43. 10.
10. Obeidat F, Ahram M, Al Khader A
et al. Clinical and histopathological features of breast cancer in
Jordan: Experience from a tertiary care hospital. J Pak Med Assoc.
2017;67(8):1206-1212.
11. Goldhirsch A, Winer EP, Coates
AS, et al. Personalizing the treatment of women with early breast
cancer:
Highlights of the st gallen international expert consensus
on the primary therapy of early breast Cancer 2013. Ann
Oncol. 2013;24(9):2206-2223.
12. Sainsbury R. The development of
endocrine therapy for women with breast cancer. Cancer Treat Rev.
2013;39(5):507-517.
13. Dao TL, Huggins C. Metastatic
cancer of the breast treated by adrenalectomy evaluation and the
five-year results Chicago Materials and Methods The first series of
patients treated ectomy , or by this operation combined
with oophor- ectomy , consisted of 52 consecutive patie.
JAMA. 1957;165(14):1793-1797.
14. Singh G. Oophorectomy in Breast
Cancer-Controversies
and Current Status. Indian J Surg. 2012;74(3):210-212.
15. Russell PW. Hypophysectomy in
metastatic breast carcinoma. J Kans Med Soc. 1955;56(9):481 485.
16. Jensen EV, Block GE, Smith S,
Kyser K DE. Estrogen
receptors and breast cancer response to adrenalectomy.
Natl Cancer Inst Monogr. 1971;34:55-.
17. Huggins C, Bergenstal DM.
Inhibition of Human Mammary and Prostatic Cancers by Adrenalectomy.
Cancer
Res. 1952;12(2):134-141.
18. Friedman N, Jaffe HL, Rabwin MH,
Rosenblum DH,
Simkin B. Adrenalectomy for control of cancer of the
breast. Calif Med. 1956;85(4):213-219.
19. Olivercrona RL and H.
Hipophysectomy in the treatment
of malignant tumors. Cancer. 1957;16(10):788-794.
20. Silverberg GD, Britt RH.
Transsphenoidal hypophysectomy in the treatment of metastatic breast
and prostate
carcinoma. West J Med. 1979;130(3):191-195.
21. Lewison EF. The treatment of
Advanced breast cancer.
Am J Nurs. 62(10):107-110.
22. Khan S. Estrogen receptor and
breast cancer: a historical perspective. Cancer Drug Discov Dev.
2019:1-14.
23. Allen E, Doisy EA. An Ovarian
Hormone: Preliminary
Report on Its Localization, Extraction and Partial Purification, and
Action in Test Animals. JAMA J Am Med Assoc. 1983;250(19):2681-2683.
24. Doisy EA. An Autobiography. Annu
Rev Biochem. 1976.
25. Herting T A. Allen and Doisy’s
“An Ovarian Hormone.”
JAMA. 1983;250(19).
26. Allen E, Doisy EA. An ovarian
hormone. Preliminary
reports on its localization, extraction and partial purification and
action in test animals. J Am Med Assoc.
1923;81(10):819-821.
27. Doisy EA. The crystals of
thefollicular ovarian hormone.
Proc Soc Exp Biol Med. 1930;27(5):417-419.
28. Butenandt A. Über “Progynon” ein
krystallisiertes
weibliches Sexualhormon. Naturwissenschaften.
1929;17(879). 29.
29. Santen RJ, Simpson E. History of
estrogen: Its purification, structure, synthesis, biologic actions, and
clinical
implications. Endocrinology. 2019;160(3):605-625.
30. Lerner LJ, Jordan VC. Development
of Antiestrogens
and Their Use in Breast Cancer: Eighth Cain Memorial
Award Lecture. Cancer Res. 1990;50(14):4177-4189.
31. Russo J, Russo IH. The role of
estrogen in the initiation of
breast cancer. J Steroid Biochem Mol Biol. 2006;102(1-5
SPEC. ISS.):89-96.
32. Haddon, A.. Influence of
synthetic oestrogens upon advanced malignant disease. Brit MedJ.
1944:2:393-398.
33. Schwander H, Marvin HN. Treatment
of carcinoma of the
human breast with testosterone propionate: A report of five
Cases. J Clin Endocrinol Metab. 1947;7(6):423-432.34.
34. Sala JM, Del Regato JA. Treatment
of carcinoma of the
endometrium. Radiology. 1962;79:12-17.
35. McGraw AB. Testosterone
propionate in treatment of recurrent cancer of the breast. Arch Surg.
1948;57(3):385-
390. 36.
36. Kennedy BJ. Massive Estrogen
Administration in premenopausal women with metastatic breast cancer.
Cancer.
1962;15(5):641-648.
37. Allen E,Doisy E. An ovarian
hormone preliminary report
on its localization, extraction and partial purification, and
action in test animals. JAMA. 1923;81(10):819.
38. Macgregor JI, Jordan VC. Basic
guide to the mechanisms
of antiestrogen action. Pharmacol Rev. 1998;50(2):151-196.
39. Craig Jordan V, McDaniel R,
Agboke F, Maximov PY.
The evolution of nonsteroidal antiestrogens to become selective
estrogen receptor modulators. Steroids.
2014;90:3-12.
40. Herbst AL, Griffiths CT, Kistner
RW. Clomiphene citrate (NSC-35770) in disseminated mammary carcinoma.
Cancer Chemother Rep. 1964;43:39-41.
41. Jensen E V. On the Mechanism of
Estrogen Action genic
activity. Perspect Biol Med .2018;6(1):47-60.
42. Jensen E V, Suzuki T, Kawashima
T, Stumpf WE, Jungblut PW, DeSombre ER. A two-step mechanism for the
interaction of estradiol with rat uterus. Proc Natl Acad
Sci. 1968;59(2):632 LP-638.
43. Moore DD. A conversation with
Elwood Jensen. Annu
Rev Physiol. 2012;74:1-11.
44. Jensen E V., Jordan VC. The
estrogen receptor:
A model for molecular medicine. Clin Cancer Res.
2003;9(6):1980-1989.
45. Gorski J, Noteboom W.
Stereospecific Binding of
Estrogens in the Rat Uterus. Arc Biochem Biophy.
1965;111(3):559-568.
46. Toft D, Shyamala G, Gorski J. A
receptor molecule for
estrogens: studies using a cell-free system. Proc Natl
Acad Sci U S A. 1967;57(6):1740-1743.
47. Greene GL, Fitch FW, Jensen E V.
Monoclonal antibodies to estrophilin: Probes for the study of estrogen
receptors. Proc Natl Acad Sci U S A. 1980;77(1):157-161.
48. King WJ, Greene GL. Monoclonal
antibodies localize
oestrogen receptor in the nuclei of target cells. Nature.
1984;307(5953):745-747.49.
49. Krust A, Green S, Argos P, et al.
The chicken oestrogen
receptor sequence: homology with v-erbA and the human oestrogen and
glucocorticoid receptors. EMBO J.
1986;5(5):891-897.
50. Kumar V, Green S, Staub A,
Chambon P. Localisation
of the oestradiol-binding and putative DNA-binding
domains of the human oestrogen receptor. EMBO J.
1986;5(9):2231-2236.
51. Williams C, Lin CY. Oestrogen
receptors in breast cancer: Basic mechanisms and clinical implications.
Ecancermedicalscience. 2013;7(1):1-13. 52.
52. Kuiper GGJM, Enmark E,
Pelto-Huikko M, Nilsson S,
Gustafsson JÅ. Cloning of a novel estrogen receptor expressed in rat
prostate and ovary. Proc Natl Acad Sci U
S A. 1996;93(12):5925-5930.
53. Todd IDH. Prediction of Response
in Cancer Therapy. Br
J Cancer. 1972;26(5):423-423.
54. Jensen E V. Hormone dependency of
breast cancer.
Cancer. 1981;47(10):2319-2326.
55. Maass H, Engel B, Hohmeister H,
Lehmann F, Trams G.
Estrogen receptors in human breast cancer tissue. Am J
Obstet Gynecol. 1972;113(3):377-382.
56. Persijn JP, Korsten CB. Oestrogen
Receptor in Human
Breast Cancer Tissue and Response to Endocrine Therapy. Br Med J.
1973;2(5869):750.
57. Criss WE, Bland KI, O’Leary JP.
Predictability of response to endocrine ablation in breast cancer.
South Med J.
1975;68(6):714-716.
58. Meredith JT, McBride RC, Cerezo
L. Estrogen receptors
in breast cancer. J Fla Med Assoc. 1988;75(1):22-28.
59. Jensen E V. Estrogen Receptors in
Hormone-Dependent Breast Cancers. Cancer Res. 1975;35:3362-3364.
60. Kaye AH, Laws ER. Historical
Perspective. Brain Tumors. 2012:1-5.
61. Kwakowsky A, Milne M, Waldvogel
H, Faull R. Effect of
Estradiol on Neurotrophin Receptors in Basal Forebrain
Cholinergic Neurons: Relevance for Alzheimer’s Disease. Int J Mol Sci.
2016;17:2122.
62. Saji S, Jensen E V, Nilsson S,
Rylander T, Warner M,
Gustafsson JA. Estrogen receptors alpha and beta in
the rodent mammary gland. Proc Natl Acad Sci U S A.
2000;97(1):337-342.
63. Platet N, Cathiard AM, Gleizes M,
Garcia M. Estrogens
and their receptors in breast cancer progression: A dual
role in cancer proliferation and invasion. Crit Rev Oncol
Hematol. 2004;51(1):55-67.
64. Saji S, Hirose M, Toi M. Clinical
significance of estrogen
receptor β in breast cancer. Cancer Chemother Pharmacol. 2005;56(SUPPL.
7):21-26.
65. Weigel NL. Steroid hormone
receptors and their regulation
by phosphorylation. Biochem J. 1996;319(3):657-667.
66. Rong C, Fasolt É, Corvin R, Hess
J. Estrogen Receptor Signaling in Radiotherapy : From Molecular
Mechanisms to Clinical Studies. 2018.
67. Katzenellenbogen BS. Estrogen
receptors: Bioactivities
and interactions with cell signaling pathways. Biol Reprod.
1996;54(2):287-293.
68. O’Malley BW. A life-long search
for the molecular
pathways of steroid hormone action. Mol Endocrinol.
2005;19(6):1402-1411. doi:10.1210/me.2004-0480.
69. Girgert R, Emons G, Gründker C.
Estrogen signaling in
ERα-negative breast cancer: ERβ and GPER. Front Endocrinol (Lausanne).
2019;10(JAN):1-12.
70. Klinge CM. Estrogens regulate
life and death in
mitochondria.J Bioenerg Biomembr 2017;49(4):307-324
71. Liao TL, Lee YC, Tzeng CR, et al.
Mitochondrial translocation of estrogen receptor β affords resistance
to oxidative insult-induced apoptosis and contributes to the
pathogenesis of endometriosis. Free Radic Biol Med.
2019;134(October 2018):359-373.
72. Vona R, Ascione B, Malorni W,
Straface E. Mitochondria
and Sex-Specific Cardiac Function. In: Advances in Experimental
Medicine and Biology. 2018:241-256.
73. Perillo B, Sasso A, Abbondanza C,
Palumbo G.
17β-Estradiol Inhibits Apoptosis in MCF-7 Cells, Inducing bcl-2
Expression via Two Estrogen-Responsive
Elements Present in the Coding Sequence. Mol Cell
Biol. 2000;20(8):2890-2901.
74. Engelbreth-Holm J. Acceleration
of the Development of
Mammary Carcinomas in Mice by Methylcholanthrene.
Cancer Res. 1941;1(2):109-112.
75. Bielschowsky F. The carcinogenic
action of 2-acetylaminofluorene and related compounds. Br Med Bull.
1946;4(5-6):382-384. 76.
76. Bielschowsky F. Distant tumours
produced by 2-aminoand 2-acetylamino-fluorene. Br J Exp Pathol.
1944;25(1).
77. Geyer RP, Bryant JE, Bleisch VR,
Peirce EM, Stare FJ.
Effect of Dose and Hormones on Tumor Production in
Rats Given Emulsified 9,10-Dimethyl-l,2-benzanthracene Intravenously.
Cancer Res. 1953;13:503-506.
78. Shay H. Development of
adenocarcinoma of the breast
in the wistar rat following the gastric instillation of
methylcholanthrene. J Natl Cancer Inst. 1949;10(2):255-266.
79. Cantarow A, Stasney J, Paschkis
KE. The lnfluence of
Sex Hormones on Mammary Tumors Induced by 2-Acetaminofluorene. Cancer
Res. 1948;8(9):412-417.
80. Huggins C, Morii S, Grand LC.
Mammary cancer induced by a single dose of polynuclear hydrocarbons:
routes of administration. Ann Surg. 1961;154(6):315-318.
81. Walsh PC. How Charles Huggins
made his nobel prize
winning discovery-in his own words: An historic audio recording.
Prostate. 2012;72(16):1718-1718.
82. Harper K, Walpole AL. A New
Derivative of triphenilethylene: effect on implatation and mode of
action in
rats. J Reprod Fert. 1966;13:101-119.
83. Klopper A, Hall M. New Synthetic
Agent for the Induction of Ovulation: Preliminary Trials in Women. Br
Med J.
1971;1(5741):152-154.
84. Jordan VC. A current view of
tamoxifen for the treatment
and prevention of breast cancer. Br J Pharmacol.
1993;119:507-517.
85. Jordan VC. Antiestrogenic and
antitumor properties
of tamoxifen in laboratory animals. Cancer Treat Rep.
1976;60(10):1409-1419.
86. Satyaswaroop PG, Zaino RJ, Mortel
R. Estrogen-like
Effects of Tamoxifen on Human Endometrial Carcinoma Transplanted into
Nude Mice. Cancer Res.
1984;44(9):4006-4010.
87. Jordan VC, Koerner S. Tamoxifen
(ICI 46,474) and the
human carcinoma 8S oestrogen receptor. Eur J Cancer.
1975;11(3):205-206.
88. Cole MP, Jones CTA, Todd IDH. A
new anti-oestrogenic
agent in late breast cancer an early clinical appraisal of
ICI46474. Br J Cancer. 1971;25(2):270-275.
89. Jordan VC. Tamoxifen: A most
unlikely pioneering medicine. Nat Rev Drug Discov. 2003;2(3):205-213.
90. Breast E, Trialists C, Group C.
Polychemotherapy for
early breast cancer: an overview of the randomised
trials. Early Breast Cancer Trialists’ Collaborative Group.
Lancet . 1998;352(9132):930-942.
91. Vogel VG, Costantino JP,
Wickerham DL, Cronin WM.
Re: Tamoxifen for prevention of breast cancer: Report of
the National Surgical Adjuvant Breast and Bowel Project
P-1 Study [1]. J Natl Cancer Inst. 2002;94(19):1504.
92. Cuzick J. First results from the
International Breast Cancer Intervention Study (IBIS-I): A randomised
prevention
trial. Lancet. 2002;360(9336):817-824.
93. Jordan VC. Tamoxifen as the first
targeted long-term adjuvant therapy for breast cancer. Endocr Relat
Cancer.
2014;21(3):1-20.
94. Santen RJ, Brodie H, Simpson ER,
Siiteri PK, Brodie
A. History of aromatase: Saga of an important biological mediator and
therapeutic target. Endocr Rev.
2009;30(4):343-375.
95. Ghosh D, Griswold J, Erman M,
Pangborn W. Structural
basis for androgen specificity and oestrogen synthesis
in human aromatase. Nature. 2009;457(7226):219-223.
96. Hemsell DL, Grodin JM, Brenner
PF, Siiteri PK, Macdonald PC. Plasma precursors of estrogen. II.
Correlation of the extent of conversion of plasma androstenedione to
estrone with age. J Clin Endocrinol Metab.
1974;38(3):476-479.
97. Grodin JM, Siiteri PK, MacDonald
PC. Source of estrogen production in postmenopausal women. Obstet
Gynecol Surv. 1973;28(9):654-656.
98. Schwarzel WC, Kruggel WG, Brodie
HJ. Studies on the
Mechanism of Estrogen Biosynthesis. VIII. The Development of Inhibitors
of the Enzyme System in Human
Placenta. Endocrinology. 1973;92(3):866-880.
99. Coombes RC, Goss P, Dowsett M,
Gazet JC, Brodie A 1984 4-Hydroxyandrostenedione in treatment of
postmenopausal patients with advanced breast cancer.
Lancet 1984;2(8414)::1237–1239.
100. Steele RE, Mellor LB, Sawyer WK,
Wasvary JM, Browne LJ 1987 In vitro and in vivo studies demonstrating
potent and selective estrogen inhibition with the nonsteroidal
aromatase inhibitor CGS 16949A. Steroids.1987;50(1-3):147–161.
101. Trunet PF, Mueller P, Girard F,
Aupetit B, Bhatnagar AS,
Zognbi F, Ezzet F, Menard J 1992 The effects of fadrozole hydrochloride
on aldosterone secretion in healthy
male subjects. J Clin Endocrinol Metab 74:571–576.
1992:1992.
102. Buzdar AU, Robertson JFR,
Eiermann W, Nabholtz JM.
An overview of the pharmacology and pharmacokinetics
of the newer generation aromatase inhibitors anastrozole, letrozole,
and exemestane. Cancer. 2002;95(9):2006-
2016. 103.
103. Janni W, Hepp P. Adjuvant
aromatase inhibitor therapy:
Outcomes and safety. Cancer Treat Rev. 2010;36(3):249-
261.
104. Behan LA, Amir E, Casper RF.
Aromatase inhibitors for
prevention of breast cancer in postmenopausal women:
A narrative review. Menopause. 2015;22(3):342-350.
105. Lai AC, Crews CM, Haven N.
Induced protein degradation. Nat Rev Drug Discov. 2017;16(2):101-114.
106. Carlson RW. The history and
mechanism of action of
fulvestrant. Clin Breast Cancer. 2005;6(SUPPL. 1):S5.
107. Robertson JFR, Jiang Z, Di Leo
A, et al. A meta-analysis of clinical benefit rates for fulvestrant 500
mg vs.
alternative endocrine therapies for hormone receptor-positive advanced
breast cancer. Breast Cancer.
2019;26(6):703-711.
108. Sutherland RL, Musgrove EA.
Cyclins and breast cancer. J Mammary Gland Biol Neoplasia.
2004;9(1):95-
104.
109. Buckley MF, Sweeney KJ, Hamilton
JA, Sini RL, Manning DL, Nicholson RI, deFazio A, Watts CK, Musgrove
EA SR. Expression and amplification of cyclin genes in
human breast cancer. Oncogene. 1993;8(8):2.
110. Asghar U, Witkiewicz AK, Turner
NC, Knudsen ES. The
history and future of targeting cyclin-dependent kinases
in cancer therapy. Nat Rev Drug Discov. 2015;14(2):130-
146.
111. Bartkova J, Lukas J, Müller H,
Lützhøt D, Strauss M,
Bartek J. Cyclin D1 protein expression and function in
human breast cancer. Int J Cancer. 1994;57(3):353-361.
112. Sedlacek HH, Czech J, Naik R, et
al. Flavopiridol (L86
8275; NSC 649890), a new kinase inhibitor for tumor
therapy. Int J Oncol. 1996;9(6):1143-1168.
113. Mita MM, Joy AA, Mita A, et al.
Randomized phase II trial
of the cyclin-dependent kinase inhibitor Dinaciclib (MK7965) vs.
capecitabine in patients with advanced breast
cancer. Clin Breast Cancer. 2014;14(3):169-176.
114. Fry DW, Harvey PJ, Keller PR, et
al. Specific inhibition of
cyclin-dependent kinase 4/6 by PD 0332991 and associated antitumor
activity in human tumor xenografts. Mol
Cancer Ther. 2004;3(11):1427-1437.
115. Toogood PL, Harvey PJ, Repine
JT, et al. Discovery of a
potent and selective inhibitor of cyclin-dependent kinase
4/6. J Med Chem. 2005;48(7):2388-2406.
116. Rader J, Russell MR, Hart LS, et
al. Dual CDK4/CDK6
inhibition induces cell-cycle arrest and senescence in
neuroblastoma. Clin Cancer Res. 2013;19(22):6173-
6182.
117. Zhang YX, Sicinska E, Czaplinski
JT, et al. Antiproliferative effects of CDK4/6 inhibition in
CDK4-amplified human liposarcoma in vitro and in vivo. Mol Cancer Ther.
2014;13(9):2184-2193
118. Gelbert LM, Cai S, Lin X, et al.
Preclinical characterization of the CDK4/6 inhibitor LY2835219: In-vivo
cell
cycle-dependent/independent anti-tumor activities alone/in combination
with gemcitabine. Invest New Drugs.
2014;32(5):825-837.
119. Tate SC, Cai S, Ajamie RT, et
al. Semi-mechanistic
pharmacokinetic/pharmacodynamic modeling of the antitumor activity of
LY2835219, a new cyclin-dependent
kinase 4/6 inhibitor, in mice bearing human tumor xenografts. Clin
Cancer Res. 2014;20(14):3763-3774.
120. Christine Desmedt CS. The most
prominent predictor of clinical outcome in breast cancer. Cell Cycle.
2006;(April):853-858.
121. Khera R, Kondamudi N, Zhong L,
Vaduganathan M, Parker J, Das SR. Temporal Trends in Heart Failure
Incidence Among Medicare Beneficiaries Across Risk Factor
Strata , 2011 to 2016. JAMA Netw. 2020;3(10):1-13.
122. Santen RJ, Manni A, Harvey H,
Redmond C. Endocrine Treatment of Breast Cancer in Women.Endocr Rev.
1990;11(2):221-65.
123. Shim W, Conaway M, Masamura S,
Yue W, Wang J,
Kmar R et al. Estradiol Hypersensitivity and MitogenActivated Protein
Kinase Expression in Long-Term Estrogen Deprived Human Breast Cancer
Cells in Vivo
.Endocrinology. 2000;2000;141(1):396-405.
124. Barone I, Cui Y, Herynk M,
Corona-Rodriguez A, Giordano C, Selever J et al. Expression of the
K303R Estrogen
Receptor α Breast Cancer Mutation Induces Resistance
to an Aromatase Inhibitor via Addiction to the PI3K/Akt
Kinase Pathway.Cancer Res. 2009;69(11):4724-4732.
125. Merenbakh-Lamin K, Ben-Baruch N,
Yeheskel A, Dvir A,
Soussan-gutman L, Jeselsohn R et al. D538G Mutation
in Estrogen Receptor- a : A Novel Mechanism for Acquired Endocrine
Resistance in Breast Cancer.Cancer Res.
2013;73(23):6856-6865.
126. Jeselsohn R, Yelensky R,
Buchwalter G, Frampton G,
Meric-Bernstam F, Gonzalez-Angulo A et al. Emergence
of Constitutively Active Estrogen Receptor- a Mutations
in Pretreated Advanced Estrogen Receptor – Positive
Breast Cancer.Clin Cancer Res. 2014;20(7):1757-1767.
127. Toy W, Shen Y, Won H, Green B,
Sakr R, Will M et al.
ESR1 ligand-binding domain mutations in hormoneresistant breast
cancer.Nat Genet 2013;45(12):1439-
1445.
128. Robinson DR, Wu YM, Vats P,Su F,
Lonigro R, Cao X
et al. Activating ESR1 mutations in hormone-resistant
metastatic breast cancer. Nat Genet. 2013;45(12):1446-
1451.
129. Harrod A, Fulton J, Nguyen VTM,
Periyasamy M, Ramos-García L, Lai C et al. Genomic modelling of the
ESR1 Y537S mutation for evaluating function and new
therapeutic approaches for metastatic breast cancer.
Oncogene. 2017;36(16):2286-2296.
130. Toy W, Weir H, Razavi P, Lawson
M, Goeppert A, Mazzola A et al. Activating ESR1 Mutations
Differentially
Affect the Effi cacy of ER Antagonists. Cancer Discov.
2017;7(3):277-287.
131. Webb PM, Cummings MC, Bain CJ,
Furnival CM. Changes in survival after breast cancer: Improvements in
diagnosis or treatment?. Breast. 2004;13(1):7-14.
132. Caswell-Jin JL, Plevritis SK,
Tian L, Cadham CX, Stout
NK, Sledge G et al. Change in Survival in Metastatic Breast Cancer with
Treatment Advances: MetaAnalysis and Systematic Review. JNCI Cancer
Spectr.
2018;2(4):1-10.
Recibido:
Octubre 27, 2020
Aprobado: Noviembre 03, 2020
Correspondencia:
Yanin Chávarri Guerra
yaninchg@gmail.com