Resúmen
Algunos de los avances más importantes en el campo de
la hematología maligna incluyen el desarrollo
de la quimioterapia, el trasplante de médula ósea y la introducción de
la terapia celular. En conjunto,
estas terapias han mejorado significativamente el pronóstico de
pacientes con enfermedades hematolinfoides.
Inicialmente el trasplante de células madre hematopoyéticas (TCMH) fue
recibido con una mezcla de
escepticismo, entusiasmo y decepciones. Inicialmente fue necesario
superar distintas barreras, incluyendo las diferencias inherentes entre
la inmunología de animales y humanos, el rechazo del injerto y
la enfermedad de injerto contra huésped (EICH). Los desenlaces médicos
y las altas tasas de mortalidad por recaída en los primeros trasplantes
frenaron, en primera instancia, la investigación del TCMH.
Sin embargo, gracias a la determinación de diferentes pioneros, el TCMH
pasó de ser una opción
experimental con disponibilidad limitada a ser una terapia que hoy en
día beneficia aproximadamente
50.000 pacientes anualmente con distintos desórdenes hematológicos que
de otro modo serían fatales.
En la actualidad el TCMH tiene una variedad de aplicaciones médicas más
allá de las neoplasias
hematológicas, incluyendo síndromes de falla medular, tratamiento de
tumores sólidos, hemoglobinopatías, enfermedades autoinmunes,
trastornos hereditarios del metabolismo e incluso enfermedades
infecciosas como el virus de inmunodeficiencia humano (VIH)(1). Además
la terapia celular, específicamente las células T con receptores de
antígeno quimérico (CAR- T) es uno de los avances más
importantes del tratamiento de las enfermedades neoplásicas
hematológica. Este artículo revisará la
perspectiva histórica del tratamiento de las neoplasias hematolinfoides
desde la quimioterapia, TCMH
y la terapia celular.
Palabras clave: Historia de la quimioterapia; trasplante
de células madre hematopoyéticas; terapia
celular; CAR- T

¹ MD. Fellow en Hematología y Oncología. Memorial Sloan Kettering
Cancer Center, División de Hematología y Oncología, Ciudad
de Nueva York, Nueva York.
² MD. Profesor Asistente. División de Hematología y Oncología Weill
Cornell Medicine, New York-Presbyterian Hospital, Ciudad de
Nueva York, Nueva York.
HISTORY OF THE TREATMENT OF HEMATOLYMPHOID
NEOPLASMS FROM CHEMOTHERAPY TO
TRANSPLANTATION AND CELL THERAPY
Abstract
Some of the most important advances in the field of
malignant hematology include the development of chemotherapy, bone
marrow transplantation, and the introduction of cell therapy.
Together, these treatment modalities have significantly improved the
prognosis of patients
with hematolymphoid diseases.
Hematopoietic stem cell transplantation (HSCT) was initially greeted
with a mixture of skepticism, enthusiasm, and disappointment.
Initially, several barriers had to be overcome, including the inherent
differences between animal and human immunology, graft rejection, and
graft-vs.-host disease (GVHD). Medical outcomes and high relapse
mortality rates in the first
transplants stopped HSCT research in the first place. However, thanks
to the determination
of different pioneers, HSCT went from being an experimental option with
limited availability
to being a therapy that today benefits approximately 50,000 patients
annually with different
hematological disorders that would otherwise be fatal.
Additionally, HSCT currently has a variety of medical applications
beyond hematologic malignancies, including bone marrow failure
syndromes, treatment of solid tumors, hemoglobinopathies, autoimmune
diseases, inherited metabolic disorders, and even infectious diseases
such
as the human immunodeficiency virus. (HIV) (1). Furthermore, cell
therapy has made recent
and impactful implications in the treatment of hematologic malignancies
of B cell origin. This
article will review the historical perspective of the treatment of
hematolymphoid neoplasms
from chemotherapy, and; HSCT and cell therapy
Keywords: History of chemotherapy; bone marrow
transplant; cell therapy; CAR-T
Historia de la quimioterapia para el
manejo de las neoplasia hematológicas
En 1845, J. H. Bennett
describió la proliferación anormal de leucocitos en sangre (2). Luego,
en 1847, R. C.
Virchow observó ciertas características en sangre que
denominó leucemia (3), del griego
leukos
cuyo significado es blanco. En 1860, cuando Biermer reportó la
incidencia alarmante de la leucemia infantil, se abrieron
las puertas para el desarrollo de medicamentos contra el
cáncer. El término quimioterapia surgió en 1909 cuando P. Ehrlich
desarrolló el medicamento para la sífilis
Salvarsan 606; la intención en ese entonces era tratar enfermedades a
partir de sustancias químicas (4).
Inadvertidamente, las guerras han llevado a desarrollos importantes en
la hematología. Los inicios del
trasplante de medula ósea surgieron a partir del proyecto Manhattan y
con ello la explosión de la bomba
atómica durante la Segunda Guerra Mundial. El ataque en el puerto de
Bari en Italia por parte de Alemania, liberó gas mostaza ocasionando
toxicidades hematológicas severas y muertes en más de mil soldados
y residentes expuestos (
Figura 1).
Posteriormente, L.
S. Goodman y A. Gilman utilizaron la mostaza nitrogenada para tratar
cánceres hematológicos como la
leucemia y el linfoma en 1946, permitiendo crear un
modelo experimental para el desarrollo de diferentes
agentes alquilantes (5).
Poco después del descubrimiento de la mostaza nitrogenada, Sidney
Farber (Figura 2) en Boston, con el
apoyo del químico Yellapragada Subbarow, demostró
que la aminopterina, un compuesto relacionado con el
ácido fólico, producía remisiones en niños con leucemia aguda al
inhibir la replicación del ADN (6). Este
medicamento fue el predecesor del metotrexato, usado
frecuentemente en la actualidad.

Figura 1. Gas mostaza. Bari, Italiza. Obtenido Mondadori, 1945
Durante las décadas de 1940 y 1950, la quimioterapia
se basó en el uso exclusivo de un único agente como pilar para el
tratamiento de la leucemia. Por ejemplo,
en 1951 G. B. Elion descubrió el efecto antitumoral
de la 6-mercaptopurina. Luego, en 1955 el Centro de
Servicio Nacional de Quimioterapia del Cáncer Estadounidense estudió a
gran escala diferentes sustancias
químicas sintéticas, productos de fermentación y derivados de plantas
como posibles agentes quimioterapéuticos. Estos esfuerzos llevaron al
desarrollo de varios tipos de fármacos como el 5-fluorouracilo, que C.
Heidelberger sintetizó en 1957, y el derivado vegetal
vincristina, desarrollado en 1958 (7).
Leucemia linfoide aguda y la introducción
de la quimioterapia combinada
En la década de1960 se introdujo la quimioterapia con
múltiples agentes, lo que permitió vencer la resistencia a agentes
únicos y así aumentar drásticamente la
supervivencia de pacientes con enfermedades hematológicas. En 1964, E.
Frei y E. Freireich utilizaron un
régimen de combinación de múltiples fármacos quimioterapéuticos en
niños con leucemia (8). La primera
quimioterapia combinada de dosis alta se denominó “VAMP”; comprendía
vincristina, ametopterina (metotrexato), mercaptopurina y prednisona.
Sin embargo, este régimen de quimioterapia de combinación de
dosis alta tuvo graves efectos adversos, lo que impulsó
ensayos clínicos adicionales para alcanzar una combinación de fármacos
con mejor perfil toxicológico
(9). En 1965 se introdujo el régimen conocido como
POMP (6-mercaptopurine vincristina, metotrexato,
y prednisona) que hoy en día continua utilizándose
como terapia de mantenimiento en la leucemia linfoblástica aguda (LLA)
(10).
No hay duda de que el tratamiento de la (LLA) ha sido
un éxito sobresaliente.. La LLA fue el primer cáncer
que entró en remisión con quimioterapia. La euforia
inicial que produjeron los resultados esperanzadores en el tratamiento
de la LLA se atenuó cuando se
evidenciaron las recaídas testiculares y en el sistema
nervioso central. Inicialmente se utilizaron la irradiación craneal y
el metotrexato intratecal. Aunque estas
modalidades son efectivas para prevenir las recaídas,
también se asociaron con toxicidades cognitivas significativas (11). En
la actualidad, en pacientes con
enfermedad de bajo riesgo y ausencia de enfermedad
residual mínima (definida como <0,01% de células
leucemia en la medula ósea) se puede evitar el uso de
irradiación craneal sin alterar las tasas de curación
(12). Hoy en día, 90% de los niños en países occidentales son
sobrevivientes a largo plazo.
La historia de la leucemia promielocítica aguda
La historia de la leucemia promielocítica
aguda
(LPMA), una variante de la leucemia mieloide aguda
(LMA), es otro avance notable en el tratamiento de la
leucemia. La LPMA se describió en 1957 por Leif K.
Hillested (13). La inducción con danorubicina no fue
exitosa y por el contrario, los pacientes fallecieron a
consecuencia de hemorragias y coagulopatías severas.
En 1981, Theodore R. Breitman indujo la diferenciación de
promielocíticos agudos de leucemia con ácido
retinoico (14). Christine Chomienne, luego demostró
los efectos diferenciadores del ácido trans retinoico
(ATRA) en células de leucemia promielocítica aguda
in vitro y, demostró que el ATRA era diez veces más
potente que el ácido 13-cis retinoico. Posteriormente,
en 1988 investigadores en Shanghái, afirmaron que la
remisión en la LPMA podría inducirse utilizando el
agente diferenciador ATRA (15).
Entre tanto, el arsénico que se utilizó en Manchuria
hace más de 2.000 años, solo se reportó en la literatura occidental en
1930 como agente terapéutico, inicialmente en el tratamiento popular
para la leucemia
mieloide crónica (LMC). El compuesto trióxido de arsénico se investigó
en China para el tratamiento contra
el cáncer desde la década de 1970 y posteriormente se
reportaron respuestas exitosas con este compuesto intravenoso en
pacientes con LPMA en 1992 (16). Ambos agentes tienen un lugar definido
en la medicina
occidental en el tratamiento de la LPMA (17, 18).
La introducción de la citogenética y el
desarrollo de Imatinib
Durante la década de 1950, los científicos tenían un
conocimiento limitado sobre la influencia de las mutaciones genéticas
en el desarrollo del cáncer. A pesar
de esto, algunos investigadores centraron sus estudios
en la relación de la citogenética y la biología del cáncer, con el fin
de identificar alteraciones cromosómicas
especificas con causalidad directa en la oncogénesis.
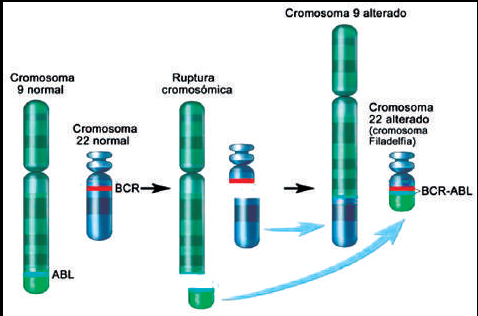
Durante ese tiempo, Nowell y Hungerford se dedicaron a entender la
biología de LMC. En 1960, estos investigadores notaron que uno de los
46 cromosomas
era anormalmente corto; éste se denominó más tarde
como “cromosoma Filadelfia” en honor a la ciudad
en donde fue descrito (19). Las investigaciones subsiguientes
demostraron que el 95% de los pacientes con
LMC tienen el cromosoma Filadelfia. Gracias al progreso en la
citogenética, se determinó que este cromosoma era el resultado de una
translocación reciproca
entre los brazos largos de los cromosomas 9 y 22 t (9,
22) (Figura 3). La translocación produce una proteína de fusión
expresada en células malignas conocida
como BCR-Abl (
breakpoint cluster
region–v-Abl Abelson
murine leukemia viral oncogene homolog) (20).
La importancia de la BCR-Abl se demostró a medida
que se caracterizaron las proteínas tirosina quinasas,
esenciales en el crecimiento y la diferenciación (21).
En consecuencia, el vínculo con la LMC se hizo evidente, al entender
que el Abl era una proteína tirosina
quinasa. Posteriormente, una serie de estudios confirmaron que la
presencia de la BCR-Abl es la causa oncogénica y no el resultado de la
LMC (22).
Con esta información, se preparó el escenario para
el diseño racional de la terapia dirigida en oncología.
Hasta entonces, la terapia estándar para la LMC era
similar al tratamiento de otras neoplasias malignas,
consistente en la administración de esquemas citotóxicos intensos,
invasivos y poco efectivos. Sin embargo,
la identificación del producto genético causante de la
enfermedad alentó a los investigadores a buscar agentes que
interfirieran específicamente con la función
de la proteína de fusión BCR-Abl. Esta búsqueda finalmente condujo a la
identificación del mesilato de
imatinib (
Gleevec) como un
inhibidor de BCR-Abl (23)
que ha afectado dramáticamente la calidad de vida de
los pacientes que padecen LMC. Así, la historia del
cromosoma Filadelfia se estableció como un nuevo paradigma donde la
observación clínica y la ciencia básica rigurosa resultaron en nuevas
hipótesis que pueden
traducirse en la práctica clínica.
Leucemia mieloide aguda y el esquema
“7+3”
Pocos enfoques terapéuticos para las enfermedades
malignas se han mantenido esencialmente iguales en
los últimos 40 años, como ocurre con la terapia de inducción para la
leucemia mieloide aguda (LMA) consistente en la infusión intravenosa
continua de arabinósido de citosina con daunorrubicina (24).
En 1951, el nucleósido natural arabinosa de timina
se aisló a partir de la esponja
Cryptotethia
crypta. A
mediados de la década de 1960, se demostró que el
clorhidrato de arabinósido de citosina inhibía el metabolismo in vitro
de los ácidos nucleicos en sistemas
celulares bacterianos, virales y tumorales (25). Schabel
y Skipper demostraron que el crecimiento de leucemia
era sensible al arabinósido de citosina durante la síntesis de ADN, la
fase S del ciclo mitótico celular (26).
Posteriormente, Freireich y sus colegas del Instituto
Nacional de Cáncer de los Estados Unidos (NCI) trataron a 14 pacientes
con LMA con una infusión continua de arabinósido de citosina durante 5
días. Seis pacientes alcanzaron una remisión parcial no duradera.
Ellison, Holland y colegas se centraron en el efecto de
la infusión continua de arabinósido de citosina sobre
el recuento de células sanguíneas al comprobar que la
frecuencia de administración continua del fármaco era
esencial para aumentar la efectividad (27).
En cuanto a la daunorrubicina, el otro pilar del esquema 7+3, éste se
aisló a principios del siglo XX a partir
de antibióticos antimitóticos de bacterias del suelo (
actinomycete). En el Hospital
General de Massachusetts,
distintos colaboradores compararon la terapia combinada con arabinósido
de citosina con 5-tioguanina,
6-mercaptopurina o daunorrubicina. Los investigadores concluyeron que
la combinación de arabinósido de
citosina con daunorrubicina fueron significativamente
superiores para inducir remisión en los pacientes con
LMA, comparado con la administración única de arabinósido de citosina
(28).
Con esta experiencia previa, en 1973, Yates y colegas
documentaron en un estudio clásico los resultados
del programa de tratamiento consistente en 7 días de
100mg /m² de arabinósido de citosina y 3 días de 45
mg/m² daunorrubicina en 16 pacientes con LMA.
63% de los pacientes entraron en remisión con este
esquema de inducción, que proporcionaba el mejor
balance entre la terapia intensiva, la probabilidad de
remisión y la toxicidad asociada a la terapia (29).
Si bien la introducción del esquema terapéutico 7+3
mejoró las tasas de remisión y supervivencia, este progreso también
surgió gracias a distintos avances en la
medicina, incluyendo el uso de catéteres centrales, la
asepsia y disponibilidad de soporte transfusional de
plaquetas. Es evidente, sin embargo, que el desarrollo de terapias
alternas de inducción es el centro de
investigación para múltiples oncólogos dedicados al
tratamiento de la LMA, que busca mejorar la supervivencia de pacientes
adultos mayores quienes con frecuencia no toleran este esquema, por
fortuna en los
últimos 5 años hay muchos nuevos tratamientos con
resultados esperanzadores (30).
Linfoma de Hodgkin
A Sir Thomas Hodgkin se le atribuye la descripción
inicial del trastorno clínico que lleva su nombre. En
1832, informó sobre un grupo de pacientes con agrandamiento de los
ganglios linfáticos y el bazo (31).
Unos 60 años después, patólogos de Alemania y Estados Unidos
describieron de forma independiente las
características microscópicas de diagnóstico del linfoma de Hodgkin.
La cura del linfoma de Hodgkin (LH) en el Siglo XX
es otra de las historias de éxito más importantes del
cáncer. Los avances en radioterapia y quimioterapia,
junto con la investigación clínica rigurosa, transformaron un trastorno
invariablemente fatal en uno curable de forma rutinaria. El impacto del
tratamiento
del LH fue, sin embargo, mucho mayor porque creó
optimismo para el tratamiento del cáncer en general
y demostró el potencial del enfoque multidisciplinario
para el diagnóstico y el tratamiento del cáncer (32).
Un equipo del Instituto Nacional del Cáncer combinó
cuatro medicamentos de quimioterapia (mostaza, vincristina,
procarbazina y prednisona) conocidos como
el régimen “MOPP” y documentó las primeras curas
del linfoma de Hodgkin avanzado en 1964 (32).
A finales de los años setenta y ochenta se presentó otro
gran avance con el régimen de quimioterapia alternativo de cuatro
fármacos (doxorrubicina, bleomicina,
vinblastina y dacarbazina), conocido como “ABVD”
que demostró ser más eficaz que el MOPP en el tratamiento de
enfermedades avanzadas y además resultaba en menor toxicidad (33).
El Grupo de Estudio de Hodgkin Alemán introdujo
un programa intensivo de quimioterapia compuesto por siete fármacos,
conocido como “BEACOPP”
(bleomicina, etoposido, doxorubicina, ciclofosfamida,
vincristina, procarbazina, y prednisona) para abordar
el hecho de que aproximadamente el 30% de pacientes
con LH avanzado no responden al ABVD en primera
línea. Si bien este régimen se asoció con una mayor
tasa de curación y supervivencia, el régimen BEACOPP también resultó en
mayor toxicidad (34).
Otros avances en los años noventa incluyeron la aplicación rutinaria de
marcadores inmunofenotipos (proteínas específicas en la superficie
celular que definen
subconjuntos de linfocitos) que mejoraron la precisión
del diagnóstico patológico y revelaron el inmunofenotipo de la célula
Reed Stenberg, característica del LH
(35). El avance más reciente en el manejo de LH ha
sido el manejo guiado por imágenes; el FDG-PET permite evaluar la
respuesta al tratamiento tempranamente para así evitar toxicidades
innecesarias (36).
Una de las lecciones más importantes fue el reconocimiento de los
efectos adversos tardíos en los sobrevivientes de LH que surgieron a
partir de la radioterapia
y la quimioterapia. Estos incluyen cánceres secundarios, enfermedades
cardíacas e infertilidad. Esto
transformó los esfuerzos de investigación centrados
en mejorar no solo las tasas de curación, sino también
disminuir las consecuencias a largo plazo.
Historia del trasplante de células
madre hematopoyéticas
Los inicios del trasplante de células madre
hematopoyéticas
La idea de extraer tejido enfermo y remplazarlo por tejido sano ha sido
un objetivo compartido por médicos
desde la antigüedad. Inicialmente se documentó en
1896 cuando se describió el uso medicinal de la médula ósea.
Posteriormente, las tragedias físicas durante la
Segunda Guerra Mundial impulsaron la investigación
del trasplante incluyendo el injerto de piel en víctimas
con quemaduras, el reconocimiento de la tipificación
del grupo sanguíneos ABO y con ello las transfusiones
de sangre.
Adicionalmente, el daño ocasionado por dosis de radiación masivas en
los supervivientes de las explosiones de bombas atómicas en Japón
estimuló el conocimiento de fallas medulares y leucemia (37). En 1949,
Jacobson y sus colegas utilizaron plomo para cubrir
los bazos de ratones sometidos a radiación corporal
total. Dos años después, Lorenz y sus colegas encontraron que la
administración de células de la médula
ósea también resultaba en protección contra la radiación (22).
Inicialmente, muchos investigadores, incluido Jacobson, teorizaron que
la protección contra
la radiación era secundaria a algún factor humoral en
el bazo o la médula. Sin embargo, a mediados de la
década de 1950, la “hipótesis humoral” fue firmemente rechazada y
remplazada por la hipótesis celular al
demostrar de manera convincente que la protección
contra la radiación surgía a partir de la presencia de
células donantes en la médula ósea (38).
Este descubrimiento fue recibido con entusiasmo
debido a las implicaciones en la biología celular y el
tratamiento de pacientes con desórdenes hematológicos. El fundamento
del TCMH era simple: las dosis
altas de radiación y/o quimioterapia destruirían la
médula enferma y suprimirían las células inmunes del
paciente para permitir el prendimiento del injerto. Si
bien Thomas y colegas demostraron que los pacientes
con leucemia tendrían una recuperación hematológica
posterior a la infusión de la célula madre, luego los
pacientes se enfrentarían al riesgo de recaída posterior
al TCMH (39).
Las barreras del trasplante de células
madre hematopoyéticas: rechazo del
injerto, enfermedad huésped contra injerto
y riesgo de recaída
En 1958, Mathé y colegas llevaron a cabo TCMH alogénico en seis
trabajadores expuestos accidentalmente
a reactores nucleares. Cuatro de los seis pacientes sobrevivieron, sin
embargo, las células del donante únicamente persistieron de manera
transitoria. En 1965,
Mathé y sus colegas trataron a un paciente con leucemia con radiación
corporal total e infusión de médula
ósea proveniente de seis familiares, sin conocimiento previo del
complejo mayor de histocompatibilidad,
HLA (
human leukocyte antigen
en inglés) (40). Si bien el
paciente entró en remisión, finalmente sucumbió a lo
que luego se denominaría enfermedad de injerto contra huésped (EICH).
Posteriormente, Bortin Mortimer et al describieron las
experiencias del TCMH llevadas a cabo entre 1939
y 1969 (41). Estos casos incluyeron 73 pacientes con
anemia aplásica, 84 con leucemia, 31 con otras enfermedades malignas
hematológicas y 15 pacientes
con síndromes de inmunodeficiencia primaria. De los
203 pacientes de trasplantes, 152 fallecieron, en 125
pacientes (60%) no hubo evidencia de prendimiento
del injerto, mientras que en 11 pacientes se encontró
quimerismo. Únicamente 3 pacientes sobrevivieron
(todos con inmunodeficiencia); el rechazo del injerto,
las infecciones, la EICH y la recurrencia de leucemia
fueron las principales causas de mortalidad.
Estos trasplantes, sin embargo, se realizaron antes del
reconocimiento de los esquemas de condicionamiento, el rol de la
histocompatibilidad y el control de la
EICH. En 1967, van Bekkum y de Vries declararon
que las fallas del TCMH ocurrieron principalmente
porque las aplicaciones clínicas se emprendieron demasiado pronto, la
mayoría sin conocimiento entre la
brecha de fisiología animal y humana. En consecuencia, el TCMH fue
declarado un fracaso por inmunólogos eminentes y un número importante
de investigadores abandonaron el campo. Afortunadamente, algunos
laboratorios continuaron diferentes estudios en animales que buscaban
comprender y superar los obstáculos
encontrados del TCMH alogénico humano(38).
Avances: control de la Enfermedad injerto
contra huésped, selección del donante y
esquemas de condicionamiento
Durante la década de los setenta, se depuró el proceso
de selección del donante, el control de EICH y los esquemas de
condicionamiento. Estos avances permitieron llevar a cabo los primeros
TCMH exitosos.
Reconocimiento y manejo de la
Enfermedad
injerto contra huésped.
Los injertos de piel estimularon el conocimiento de
la tolerancia inmunológica y alo-reactividad. Gorer y
Snell, identificaron el complejo mayor de histocompatibilidad (CMH) en
ratones (42) y, con este antecedente, se reconoció que el rechazo del
injerto era un fenómeno inmunológico relacionado con los antígenos
de histocompatibilidad (43). El grupo de Van Bekkum
en Holanda utilizó primates, George Santos en Johns
Hopkins (44) eligió ratas y el grupo de Seattle eligió
perros consanguíneos como modelos experimentales.
Los perros resultaron ser un modelo animal útil puesto
que estos albergan una diversidad genética amplia y
compartían enfermedades espontáneas con los humanos, como el linfoma no
Hodgkin y SCID ligado al
cromosoma X (45).
La tipificación del sistema de histocompatibilidad de
perros también permitió explorar a mayor profundidad la EICH. Los
perros que recibieron injertos con
antígeno leucocitario de perro (DLA, dog leukocyte
antigen) compatibles de la misma camada o no relacionados sobrevivieron
significativamente más tiempo
comparados con el grupo control con incompatibilidad del DLA (46). Si
bien la EICH grave se describió
por primera vez en ratones con incompatibilidad H-2,
los estudios en caninos llamaron la atención sobre la
EICH fatal y el rol de los antígenos del complejo de
histocompatibilidad menor.
Además, los estudios en
caninos eventualmente condujeron a formas de comprender, prevenir y
superar la sensibilización inducida
por transfusiones.
Al confirmar que el grado de histocompatibilidad era
esencial para reducir tanto el rechazo como la EICH
se describieron los primeros antígenos HLA en humanos (47), (48).
Posteriormente, se identificó a la EICH
crónica como un problema adicional en los sobrevivientes a largo plazo.
El control de la EICH aguda y
crónica se hizo posible al combinar el metotrexato con
inhibidores de calcineurina como la ciclosporina y el
tacrolimus. Las combinaciones de drogas inmunoreguladoras siguen siendo
un pilar en la prevención de
la EICH.
Fuentes de célula madre
Únicamente alrededor del 25-35% de pacientes tienen
hermanos con HLA idéntico. Por lo tanto, se han explorado fuentes
alternativas de donantes compatibles
con HLA incluyendo voluntarios no relacionados.
Para ampliar el grupo de donantes, se establecieron
registros que actualmente incluyen a más de 30 millones de voluntarios
no emparentados. La probabilidad
de encontrar donantes no emparentados adecuados
es aproximadamente 80% para pacientes caucásicos,
aunque este porcentaje disminuye drásticamente para
poblaciones minoritarias o con ascendencia étnica heterogénea (
Figura 4) (49)
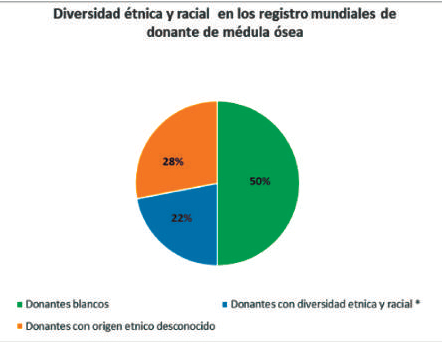
Figura 4. Grupos con diversidad étnica/racial incluyen
hispanos o latinos, afrodescendientes, asiáticos e indios
americanos. Fuente: National Marrow Donor Program,
be the Match 2016.
Una fuente de célula madre alternativa es la sangre
de cordón umbilical, que no requiere compatibilidad
absoluta del HLA y ha dado lugar a resultados alentadores en pacientes
con enfermedades hematológicas malignas. Las ventajas de esta terapia
incluyen su
disponibilidad, tasas menores de EICH y la oportunidad de trasplantar a
pacientes de minorías étnicas no
representados en los registros de donantes. Sin embargo esta modalidad
de trasplante se asocia con una
demora en la recuperación hematológica (50).
Los métodos iniciales de depleción de células T incluyeron la
centrifugación de contraflujo y fraccionamiento en gradientes de
densidad (51). En 1981, se
utilizó la globulina antitimocitos (ATG, por sus siglas
en inglés) y los anticuerpos monoclonales para prevenir la EICH. Sin
embargo, el agotamiento de las células T también condujo a diferentes
complicaciones,
entre ellas un aumento del rechazo al injerto, reconstitución inmune
retardada, mayor riesgo enfermedad
linfoproliferativa asociada al virus de Epstein-Bar y
reactivación del citomegalovirus (CMV). Inicialmente
la supervivencia global no mejoró significativamente
en comparación con la médula ósea sin depleción de
células T (52).
La identificación de la glicoproteína CD34 permitió
llevar a cabo el aislamiento de células progenitoras
hematopoyéticas, proporcionando otra fuente viable
de células madre en sangre periférica (53). Además,
factores de crecimiento tales como el factor estimulante de colonias de
granulocitos (filgrastim) y posteriormente el plerixafor (una molécula
que inhibe la
unión del receptor de quimiocinas CXCR4 al factor 1
derivado de células estromales), permitieron movilizar las células
CD34+ a la circulación periférica y por
lo tanto la posibilidad de utilizarlas como fuente de
células madre hematopoyéticas. Esta modalidad permite utilizar altas
dosis de quimioterapia como terapia de rescate en pacientes con
neoplasias resistentes
al tratamiento previo al TCHM, lo que resultó en un aumento de
supervivencia al disminuir la progresión
tumoral (54).
Efecto injerto contra leucemia
En pacientes con leucemia y otras neoplasias hematológicas, la recaída
de la enfermedad después del TCM
sigue siendo un problema importante. Si bien aumentar la intensidad del
condicionamiento reduce el riesgo
de recaída, en paralelo se aumenta la mortalidad independiente a la
recaída. Weiden y el grupo de Seattle
describieron, entre 1979 y 1981, el efecto de injerto
contra leucemia (GvL,
graft vs.
leukemia) en humanos
(55). Posteriormente se introdujo la infusión de linfocitos del donante
(DLI) para combatir la recaída (56).
Esquemas de condicionamiento
Los primeros trasplantes exitosos se llevaron a cabo
en pacientes con trastornos de inmunodeficiencia primaria, incluyeron
la agamaglobulinemia tímica (57) y
el síndrome de Wiskott-Aldrich, que fue exitoso luego
de la introducción de la ciclofosfamida (Cy) la cual, a
su vez, permitió la recuperación completa de células
T y B (58). Durante los primeros 7 u 8 años, la mayoría de los casos de
TCMH ocurrieron en pacientes
con enfermedades hematológicas avanzadas y aplasia
anémica. Estos pacientes requieren múltiples soportes
transfusionales y profilaxis o tratamiento de infecciones bacterianas,
fúngicas y virales. En consecuencia,
el desarrollo de este campo también produjo avances
importantes en conocimiento de la medicina transfusional y de las
enfermedades infecciosas.
La segunda mitad de la década de 1960 vio el refinamiento de esquemas
de condicionamiento de alta intensidad, que incluyeron la irradiación
corporal total
(ICT) fraccionada y la introducción de nuevos fármacos mieloablativos o
inmunosupresores, incluyendo la
Cy y el busulfan (Bu) (59). Estos esquemas mejoraron
el prendimiento del injerto y resultaron en muerte tumoral, de forma
semejante que la ICT. Sin embargo,
los esquemas de condicionamiento intensos son riesgosos y por lo tanto
generalmente restringidos a pacientes jóvenes o adultos sin
comorbilidades significativas. Para permitir la inclusión de pacientes
mayores,
quienes son el grupo poblacional con mayor prevalencia de neoplasias
hematológicas, se han desarrollado
programas de condicionamiento menos intensivos y
con dosis menores de ICT.
Historia de las células T con
receptores de antígeno quimérico
Las células T con receptores de antígeno quimérico
(CAR-T, por sus siglas en inglés) son linfocitos T que
se recolectan de un paciente y son genéticamente alterados en el
laboratorio para expresar un receptor modificado compuesto por dos
elementos: el componente
extracelular que funciona con la especificidad de un
anticuerpo para reconocer un antígeno particular en la
superficie de las células cancerígenas [Anticuerpo de
cadena sencilla (scFV)] , y un componente intracelular , el receptor de
célula T (TCR cc, por sus siglas en
ingles “T cell receptor”) que activa la célula T (Figura
5). Cuando el anticuerpo de cadena sencilla encuentra
el antígeno al cual está dirigido, se activa el TCR cc
que a su vez inicia una cascada de activación del linfocito, sin
necesidad de la coestimulación del complejo
mayor de histocompatibilidad (CMH), esto genera la
producción de citosinas, activa la señal para proliferación y expansión
de las células CAR-T y causa la
citotoxicidad directa contra el tumor (i.e perforinas y
eje FAS/FAS ligando).
Las investigaciones que resultaron en la producción de
las células de CAR-T llevan más de 60 años e iniciaron
con el descubrimiento de las células T. En 1961, el inmunólogo Jacques
Miller estudiaba el timo durante su
PhD en la Universidad de Londres, cuando descubrió
que este órgano era responsable de producir linfocitos
con características particulares para la defensa contra infecciones que
luego nombró “linfocitos derivadas
del Timo”(60), y hoy conocemos como Linfocitos T.
Los linfocitos T son células que se producen en la medula ósea que
migran hacia el timo donde maduran y
adquieren el TCR c en su membrana que permite reconocer antígenos
(p.ej. péptidos de virus o bacterias)
presentados por células del sistema inmune usando la
interacción con el CMH en su membrana. La interacción entre el CMH, el
TCRcc, el péptido presentado,
y las señales de coestimulación producen la estimulación que ocasiona
la activación de los linfocitos T.
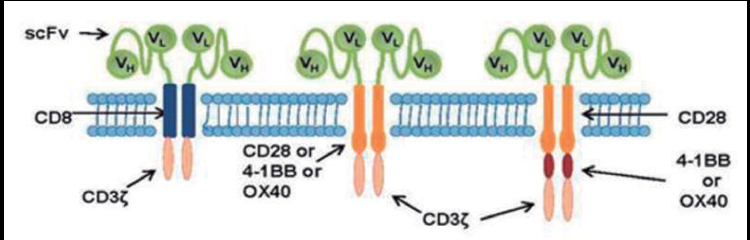
Figura 5. Estructura de las células T con receptores de antígeno
quimérico. Los CAR-T de primera generación están
compuestos por un fragmento de anticuerpo (scFV) que contiene cadena
pesada (VH) y liviana (VL), fusionando a
una región trasnmembrana de linfocitos CD8 que a su vez están
fusionando al receptor de la célula T (más común
TCR cc). La segunda generación incluye señales de coestimulación (CD28,
4-1BB o OX40). La tercera generación
contiene dos señales de coestimulación en tándem. Obtenido de PArk,
J.H. & Brentjens, R. J. (2010). Adoptive
immunotherapy for B-cell malignancies with autologous chimeric antigen
receptor modified tumor targeted T cells.
Discovery medicine, 9(47), 277-288.
Uno de los mayores avances en el conocimiento de
las implicaciones de las células T en la inmunología
tumoral surgió en 1986 cuando Steven Rosenberg
publicó una investigación liderada en NIH, que demostró que las células
T que infiltran algunos tumores podían aislarse a partir de una biopsia
del tumor,
expandirse en el laboratorio y curar algunos tumores al ser
administradas al paciente con interleucinas
(61). Este descubrimiento demostró que el sistema
inmunológico, liderado por las células T, podría jugar un papel
importante en la inmunoterapia contra
el cáncer.
Los avances en la ingeniería genética en la década de
los noventa hicieron posible esta idea. Uno de los lideres principales,
el Dr. Michel Sadelain, durante su tesis
doctoral en el Instituto de tecnología de Massachusetts
(MIT), demostró que era posible usar un vector como
un virus retroviral o un lentivirus para introducir un
nuevo gen en los linfocitos y manipular la expresión
de este. Se demostró que era posible insertar una nueva proteína en el
linfocito, como un receptor de membrana usando esta técnica. El código
de la proteína es
una secuencia de ARN, que se empaca en plásmidos
y se inserta en el retrovirus y lentivirus modificados.
Después de que el virus infecta una célula y libera su
material genético en ella, la cadena de ARN se transcribe por las
enzimas del virus generalmente usadas
para su replicación en una cadena de ADN. Este ADN
se puede incorporar en el ADN del linfocito donde se
inicia la trascripción y traducción de la proteína deseada; en el caso
de células CAR-T un nuevo receptor de
membrana con los dos componentes extracelulares e
intracelulares descritos (scFV –TCR).
La primera célula T con receptor quimérico fue producida por los
inmunólogos Zelig Eshhary Gideon Gross en 1993 en el Instituto de
Ciencia Weizmann en Israel
al insertar un receptor quimérico en el linfocito con
una parte de un anticuerpo y fusionarla al receptor de
la célula T (scFV -TCR) (62). Aunque no se pudo demostrar eficacia ni
persistencia en los modelos animales de la primera generación de
células CAR-T, estas
en todo caso representan el inicio de esta revolución.
Para mejorar el modelo, era necesario superar dos desafíos principales:
primero cómo activar y expandir
las células CAR-T en el laboratorio independiente de
células dendríticas y segundo, cómo mejorar la coestimulación de las
células CAR-T para aumentar su
activación y proliferación dentro del organismo. Los
primeros avances en el primer desafío se dieron al descubrir una nueva
técnica para hacer cultivos celulares
de linfocitos T infectados por el HIV. Carl H. June y
Bruce Levine, que iniciaron sus estudios en el Instituto
Naval de Investigación de Medicina en EEUU y continuaron en la
Universidad de Pensilvania, usaron una
técnica con microperlas diminutas donde adicionaron
en la superficie dos proteínas que imitaban moléculas
de las células dendríticas (CD3/CD28). Las células
T al estar en contacto con las microperlas se activan
y expanden, generando millones de copias que pueden permanecer vivas en
cultivos celulares (63). Estos
resultados reportados en 1996 fueron fundamentales
para establecer los procesos de manufacturación de células CAR-T.
Por otro lado, para mejorar la activación y proliferación
dentro del organismo se tuvo que investigar el papel que
juegan las señales de coestimulación. Los CAR-T de
primera generación tenían solo CD3 ζ que resultó ser
insuficiente para activar la célula T. En 1998, el Dr. Michel Sadelain
en su laboratorio del Memorial Sloan Kettering (MSKCC) publicó la
eficacia del receptor CD28
para permitir la activación de células T y aumentar la
proliferación (64). En el 2002, el laboratorio del Dr. Levine confirmó
estos resultados y publicó sus estudios
usando 4-1BB (65). Con esto se inició la creación de los
CAR-T de segunda generación que poseen una co-estimulación interna (más
frecuentemente CD28 o 4-1BB)
fusionado a CD3 ζ. En el 2002 se desarrolló el primer
modelo de segunda generación que mostró eficacia en
célula CAR-T contra antígenos prostáticos por el equipo en MSKCC en
Nueva York compuesto por Michel
Sadelin, Renier Brentjens e Isabelle Rivière.
En el 2003, el grupo de MSKCC, publicó el primer
modelo de ratones usando células CAR-T de segunda generación dirigidos
contra el antígeno CD19 - con
CD28 que podían ocasionar muerte celular de tumores
originados en células B (66). Utilizar el antígeno CD19
resultó ser un blanco ideal por su frecuencia y alta expresión en
tumores de células B. Además, el CD19 es
requerido para el desarrollo normal de células B y no
se expresa en otras células. Estos estudios preclínicos
permitieron conducir ensayos clínicos utilizando este
antígeno.
En el 2009, el equipo de MSKCC publicó su evidencia y validación de
manufacturación de CAR-T CD19
(con coestimulación de CD28) para humanos y anunció el inicio de
estudios clínicos fase 1 para leucemia
linfocítica crónica y leucemia linfoblástica aguda de
célula B (67). Simultáneamente el equipo de la Universidad de
Pensilvania con Carl June y David Porter
iniciaron el primer ensayo clínico utilizando CAR-T
contra CD19 (4-1BBB) en 3 pacientes con leucemia
linfocítica crónica y en 2011 hacen el primer los primeros reportes con
dos remisiones completas y una
parcial (68),(69).
Estos estudios ofrecieron la evidencia necesaria para
iniciar otros estudios de fase 1. Emily Whitehead, fue
una de las primeras pacientes en el ensayo clínico del
grupo de la Universidad de Pensilvania que incluyó pacientes con LLA
tipo B con células CAR-T (4-1BBB)
para pacientes pediátricos. Su caso en el 2011 es mundialmente conocido
porque después de la tercera infusión desarrolló hipertermia severa y
coma, la causa en este entonces se desconocía. Se demostraron niveles
de
interleucina-6 extremadamente altos y el Dr. June decidió tratarla con
tocilizumab, un anticuerpo contra el
receptor de la IL-6, con el cual estaba familiarizado ya
que su hija sufría de artritis reumatoide y era tratada
con este medicamento. Después de la primera dosis,
Emily Whitehead rápidamente respondió y despertó
para su cumpleaños número 7. Con este caso se empezó a reconocer los
dos efectos adversos principales
de las células de CAR-T ; el síndrome de liberación de
citocinas (
CRS por sus siglas en
inglés,
Cytokine release
syndrome) y la neurotoxicidad (70). Poco después el grupo
de MSKCC, liderado por el Dr. Brentjens y Sadelain,
publicaron su estudio clínico de CAR-T (CD19-CD28)
para tratar la LLA de célula B en adultos (71).
Después de estos reportes, la revista Science anunció la
inmunoterapia tumoral como el descubrimiento del año
y la FDA designó un estatus de “innovación” a la investigación clínica
dándole todo el soporte para acelerar
el desarrollo de estas terapias. Esto generó un auge de
estudio clínicos y, finalmente en el 2017 la FDA aprobó
las células CAR-T de CD19. El producto de Novartis,
desarrollado en colaboración con el grupo del Dr. June
(Tisagenlecleucel-Kymriah), es CD19 con coestimulación de 4-1BB-CD3
para LLA refractaria o en recaída
(menores de 25 años) y para linfoma difuso de células B. El otro
producto de Kite/Gilead (Axicabtagene
Ciloleucel-Yescarta), es CD19 con co-estimulación de
CD28-CD3 ζ para linfoma difuso de células B.
Esta tecnología ha abierto nuevas oportunidades de
tratamiento para pacientes que en su ausencia tenían
muy pocas opciones terapéuticas y pronósticos inciertos. En pacientes
pediátricos, el 81% de los 75 pacientes con LLA en recaída o
refractaria que fueron tratados en el ensayo clínico de fase 2 con
Tisagenlecleucel,
alcanzaron una respuesta completa (72). La supervivencia libre de
progresión al año fue de 50% y la supervivencia global fue del 76%.
Aunque el CRS severo
(grado III/IV) ocurrió en el 73% de los pacientes, se
demostró que el tocilizumab era efectivo. Los eventos
neurológicos ocurrieron en el 40% de los pacientes y se
trataron con cuidados de soporte.
Por otro lado, los pacientes adultos con linfoma difuso de células B en
caída o refractario, tratados en los
ensayos clínicos con Tisagenlecleucel o Axicabtagene ciloleucel han
mostrado respuestas completas en
alrededor del 50% y la supervivencia media libre de
progresión para estos pacientes no se ha alcanzado. El
CRS severo ocurre en menos del 20% de los pacientes
y la neurotoxicidad ocurre en alrededor del 10% (73).
Actualmente la FDA está próxima a aceptar los nuevos CAR-T dirigidos al
BCMA (antígeno de maduración de las células B) para pacientes con
mieloma
múltiple. En este momento hay cientos de ensayos
clínicos dirigidos a múltiples antígenos para diferentes cánceres
incluyendo tumores sólidos (p.ej. CD22,
ERBB2/HER2, EGFR, entre muchos otros). Incluso,
nuevos estudios están utilizando CAR-T generados en
donantes sanos (CAR-T alogénico). La terapia celular
ha avanzado significativamente en la última década y
esperamos en los próximos años llevar nuevas opciones de tratamiento
personalizado a los pacientes. En
todo caso, el mayor desafío actual es poder disminuir
el gasto de producción y llevar estas tecnologías mundialmente.
Conclusión
No hay duda de que los avances ocurridos en el siglo XX y en lo que va
del XXI en las enfermedades
hematolinfoides han mejorado significativamente la
supervivencia de pacientes que, de otra forma, fallecerían
prematuramente. Sin embargo, queda mucho por
avanzar para ofrecer mejores esquemas de tratamiento
que, integrando el conocimiento molecular, la inmunoterapia y la
terapia dirigida, resulten no solamente
en el aumento de la supervivencia, sino también en la
mejora de la calidad de vida de los pacientes.
Referencias
1. Hütter G, Nowak D, Mossner M,
Ganepola S, Müssig A,
Allers K et al. Long-term control of HIV by CCR5 Delta32/Delta32
stem-cell transplantation. N Engl J Med.
2009;360(7):692-8.
2. Bennett JH. Case of hypertrophy of
the spleen and liver
in which death took place from suppuration of the blood.
Edinb Med Surg J. 1845;64,:413±23.
3. Virchow R. Weisses Blut und
Milztumoren. Medicale
Zeitung. 1847;16.
4. Thorburn AL. Paul Ehrlich: pioneer
of chemotherapy and cure by arsenic (1854-1915). Br J Vener Dis.
1983;59(6):404-5.
5. Goodman LS, Wintrobe MM et al.
Nitrogen mustard therapy; use of methyl-bis (beta-chloroethyl) amine
hydrochloride and tris (beta-chloroethyl) amine hydrochloride
for Hodgkin's disease, lymphosarcoma, leukemia and
certain allied and miscellaneous disorders. J Am Med
Assoc. 1946;132:126-32.
6. Farber S, Diamond LK. Temporary
remissions in acute leukemia in children produced by folic acid
antagonist, 4-aminopteroyl-glutamic acid. N Engl J Med.
1948;238(23):787-93.
7. Pui CH, Evans WE. A 50-year
journey to cure childhood acute lymphoblastic leukemia. Semin Hematol.
2013;50(3):185-96.
8. Freireich EJ, Gehan E, Frei E,
Iii, Schroeder LR, Wolman IJ, Anbari R et al. The Effect of
6-Mercaptopurine
on the Duration of Steroid-induced Remissions in Acute
Leukemia: A Model for Evaluation of Other Potentially
Useful Therapy. Blood. 1963;21(6):699-716.
9. Frei 3rd E, Karon M, Levin RH,
Freireich EJ, Taylor
RJ, Hananian J et al. The effectiveness of combinations of antileukemic
agents in inducing and maintaining remission in children with acute
leukemia. Blood.
1965;26(5):642-56.
10. Rodriguez V, Hart JS, Freireich
EJ, Bodey GP, McCredie
KB, Whitecar JR. JP et al. POMP combination chemotherapy of adult acute
leukemia. Cancer. 1973;32(1):69-75.
11. Piller GJ. Leukaemia – a brief
historical review from ancient times to 1950. Br J Haematol.
2001;112(2):282-92.
12. Smith MA, Altekruse SF, Adamson
PC, Reaman GH,
Seibel NL. Declining childhood and adolescent cancer
mortality. Cancer. 2014;120(16):2497-506.
13. Hillestad LK. Acute Promyelocytc
Leukemia. Acta Med
Scand. 1957;159(3):189-94.
14. Breitman T, Collins S, Keene B.
Terminal differentiation
of human promyelocytic leukemic cells in primary culture
in response to retinoic acid. Blood. 1981;57(6):1000-4.
15. Degos L, Wang ZY. All trans
retinoic acid in acute promyelocytic leukemia. Oncogene.
2001;20(49):7140-5.
16. Antman KH. Introduction: the
history of arsenic trioxide
in cancer therapy. Oncologist. 2001;6 Suppl 2:1-2.
17. Bruserud O, Gjertsen BT, Huang T.
Induction of differentiation and apoptosis- a possible strategy in the
treatment of adult acute myelogenous leukemia. Oncologist.
2000;5(6):454-62.
18. Wang Z, Sun G, Shen Z, Chen S,
Chen Z. Differentiation
therapy for acute promyelocytic leukemia with all-trans
retinoic acid: 10-year experience of its clinical application. Chin Med
J (Engl). 1999;112(11):963-7.
19. Nowell PC. The minute chromosome
(Phl) in chronic
granulocytic leukemia. Blut. 1962;8:65-6.
20. Wong S, Witte ON. The BCR-ABL
story: bench to bedside and back. Annu Rev Immunol. 2004;22:247-306.
21. Goldman JM. Chronic myeloid
leukemia: a historical
perspective. Semin Hematol. 2010;47(4):302-11.
22. McNicholl F. a history of
haematology. from herodotus to hiv (oxford medical histories). Ulster
Med J.
2017;86(1):50.
23. Konopka JB, Watanabe SM, Witte
ON. An alteration of the human c-abl protein in K562 leukemia cells
unmasks associated tyrosine kinase activity. Cell.
1984;37(3):1035-42.
24. Lichtman MA. A historical
perspective on the development of the cytarabine (7days) and
daunorubicin (3days)
treatment regimen for acute myelogenous leukemia:
2013 the 40th anniversary of 7+3. Blood Cells Mol Dis.
2013;50(2):119-30.
25. Evans JS, Musser EA, Bostwick L,
Mengel GD. The
Effect of 1-β-D-Arabinofuranosylcytosine Hydrochloride
on Murine Neoplasms. Cancer Res. 1964;24(7):1285-
93.
26. Schabel FM, Johnston TP, McCaleb
GS, Montgomery
JA, Laster WR, Skipper HE. Experimental Evaluation
of Potential Anticancer Agents. VIII Effects of Certain Nitrosoureas on
Intracerebral L1210 Leukemia.
1963;23(5):725-33.
27. Freireich EJ, Wiernik PH,
Steensma DP. The Leukemias: A Half-Century of Discovery. J Clin Oncol.
2014;32(31):3463-9.
28. Carey RW, Ribas-Mundo M, Ellison
RR, Glidewell O,
Lee ST, Cuttner J et al. Comparative study of cytosine
arabinoside therapy alone and combined with thioguanine,
mercaptopurine, or daunorubicin in acute myelocytic
leukemia. Cancer. 1975;36(5):1560-6.
29. Yates JW, Wallace HJ, Jr.,
Ellison RR, Holland JF. Cytosine arabinoside (NSC-63878) and
daunorubicin (NSC83142) therapy in acute nonlymphocytic leukemia.
Cancer Chemother Rep. 1973;57(4):485-8.
30. Winer ES, Stone RM. Novel therapy
in Acute myeloid
leukemia (AML): moving toward targeted approaches.
Ther Adv Hematol. 2019;10:2040620719860645.
31. Hodgkin. On some Morbid
Appearances of the Absorbent
Glands and Spleen. Med Chir Trans. 1832;17:68-114.
32. Bonadonna G, Zucali R, Monfardini
S, De Lena M, Uslenghi C. Combination chemotherapy of Hodgkin's disease
with adriamycin, bleomycin, vinblastine, and imidazole carboxamide vs.
MOPP. Cancer. 1975;36(1):252-9.
33. Bonadonna G. Chemotherapy
strategies to improve
the control of Hodgkin's disease: the Richard and Hinda Rosenthal
Foundation Award Lecture. Cancer Res.
1982;42(11):4309-20.
34. Diehl V, Franklin J, Pfreundschuh
M, Lathan B, Paulus
U, Hasenclever D et al. Standard and Increased-Dose
BEACOPP Chemotherapy Compared with COPP-ABVD
for Advanced Hodgkin's Disease. New England Journal
of Medicine. 2003;348(24):2386-95.
35. Roullet MR, Bagg A. Recent
insights into the biology of
Hodgkin lymphoma: unraveling the mysteries of the Reed-Sternberg cell.
Expert Rev Mol Diagn. 2007;7(6):805-
20.
36. Johnson P, Federico M, Kirkwood
A, Fosså A, Berkahn
L, Carella A et al. Adapted Treatment Guided by Interim PET-CT Scan in
Advanced Hodgkin's Lymphoma. N
Engl J Med. 2016;374(25):2419-29.
37. Smith LH. Radiation Chimaeras.D.
W. van Bekkum and
M. J. de Vries. Academic Press, New York, 1967.x + 277
pp., illus. $20. Science. 1968;159(3812):294-5.
38. de la Morena MT, Gatti RA. A
history of bone marrow transplantation. Immunol Allergy Clin North Am.
2010;30(1):1-15.
39. Thomas ED, Storb R, Clift RA,
Fefer A, Johnson L, Neiman PE et al. Bone-marrow transplantation
(second of
two parts). N Engl J Med. 1975;292(17):895-902.
40. Mathé G, Amiel JL, Schwarzenberg
L, Cattan A, Schneider M. Adoptive immunotherapy of acute leukemia:
experimental and clinical results. Cancer Res.
1965;25(9):1525-31.
41. Bortin MM. A compendium of
reported human bone marrow transplants. Transplantation.
1970;9(6):571-87.
42. Snell GD, Stevens LC.
Histocompatibility genes of mice.
III. H-1 and H-4, two histocompatibility loci in the first
linkage group. Immunology. 1961;4(4):366-79.
43. Billingham RE, Brent L, Medawar
PB. ‘Actively Acquired
Tolerance’ of Foreign Cells. Nature. 1953;172(4379):603-
6.
44. Santos GW, Tutschka PJ,
Brookmeyer R, Saral R, Beschorner WE, Bias WB et al. Marrow
Transplantation
for Acute Nonlymphocytic Leukemia after Treatment
with Busulfan and Cyclophosphamide. N Engl J Med.
1983;309(22):1347-53.
45. Buckley RH. A historical review
of bone marrow transplantation for immunodeficiencies. J Allergy Clin
Immunol. 2004;113(4):793-800.
46. Epstein RB, Storb R, Ragde H,
Thomas ED. Cytotoxic
typing antisera for marrow grafting in littermate dogs.
Transplantation. 1968;6(1):45-58.
47. Dausset J. [Presence of A & B
antigens in leukocytes
disclosed by agglutination tests]. C R Seances Soc Biol
Fil. 1954;148(19-20):1607-8.
48. Ceppellini R, van Rood JJ. The
HL-A system. I. Genetics
and molecular biology. Semin Hematol. 1974;11(3):233-
51.
49. Aljurf M, Weisdorf D, Alfraih F,
Szer J, Müller C, Confer D et al. “Worldwide Network for Blood &
Marrow
Transplantation (WBMT) special article, challenges facing emerging
alternate donor registries”. Bone Marrow
Transplant. 2019;54(8):1179-88.
50. Gluckman E, Rocha V,
Boyer-Chammard A, Locatelli F,
Arcese W, Pasquini R et al. Outcome of cord-blood transplantation from
related and unrelated donors. Eurocord
Transplant Group and the European Blood and Marrow
Transplantation Group. N Engl J Med. 1997;337(6):373-
81.
51. de Witte T, Hoogenhout J, de Pauw
B, Holdrinet R, Janssen J, Wessels J et al. Depletion of donor
lymphocytes
by counterflow centrifugation successfully prevents acute
graft-vs.-host disease in matched allogeneic marrow
transplantation. Blood. 1986;67(5):1302-8.
52. Daniele N, Scerpa MC, Caniglia M,
Ciammetti C, Rossi
C, Bernardo ME, et al. Overview of T-cell depletion in
haploidentical stem cell transplantation. Blood Transfus.
2012;10(3):264-72.
53. Krause DS, Fackler MJ, Civin CI,
May WS. CD34: structure, biology, and clinical utility. Blood.
1996;87(1):1-13.
54. Kessinger A. Utilization of
Peripheral Blood Stem Cells
in Autotransplantation. Hematol Oncol Clin North Am.
1993;7(3):535-45.
55. Weiden PL, Flournoy N, Thomas ED,
Prentice R, Fefer
A, Buckner CD et al. Antileukemic effect of graft-vs.-host
disease in human recipients of allogeneic-marrow grafts.
N Engl J Med. 1979;300(19):1068-73.
56. Kolb HJ, Mittermüller J, Clemm C,
Holler E, Ledderose G, Brehm G et al. Donor leukocyte transfusions for
treatment of recurrent chronic myelogenous leukemia in
marrow transplant patients. Blood. 1990;76(12):2462-5.
57. Gatti RA, Meuwissen HJ, Allen HD,
Hong R, Good RA.
Immunological reconstitution of sex-linked lymphopenic
immunological deficiency. Lancet. 1968;2(7583):1366-9.
58. Bach FH, Albertini RJ, Joo P,
Anderson JL, Bortin MM.
Bone-marrow transplantation in a patient with the Wiskott-Aldrich
syndrome. Lancet. 1968;2(7583):1364-6.
59. Santos GW. Preparative regimens:
chemotherapy vs.
chemoradiotherapy. A historical perspective. Ann N Y
Acad Sci. 1995;770:1-7.
60. Miller JF. Immunological function
of the thymus. Lancet.
1961;2(7205):748-9.
61. Rosenberg SA, Spiess P,
Lafreniere R. A new approach
to the adoptive immunotherapy of cancer with tumorinfiltrating
lymphocytes. Science. 1986;233(4770):1318-
21.
62. Eshhar Z, Waks T, Gross G,
Schindler DG. Specific activation and targeting of cytotoxic
lymphocytes through
chimeric single chains consisting of antibody-binding
domains and the gamma or zeta subunits of the immunoglobulin and T-cell
receptors. Proc Natl Acad Sci U S
A. 1993;90(2):720-4.
63. Levine BL, Cotte J, Small CC,
Carroll RG, Riley JL,
Bernstein WB, et al. Large-scale production of CD4+ T
cells from HIV-1-infected donors after CD3/CD28 costimulation. J
Hematother. 1998;7(5):437-48.
64. Krause A, Guo HF, Latouche JB,
Tan C, Cheung NK,
Sadelain M. Antigen-dependent CD28 signaling selectively enhances
survival and proliferation in genetically
modified activated human primary T lymphocytes. J Exp
Med. 1998;188(4):619-26.
65. Maus MV, Thomas AK, Leonard DG,
Allman D, Addya
K, Schlienger K, et al. Ex vivo expansion of polyclonal
and antigen-specific cytotoxic T lymphocytes by artificial
APCs expressing ligands for the T-cell receptor, CD28
and 4-1BB. Nat Biotechnol. 2002;20(2):143-8.
66. Brentjens RJ, Latouche JB, Santos
E, Marti F, Gong
MC, Lyddane C, et al. Eradication of systemic B-cell
tumors by genetically targeted human T lymphocytes
co-stimulated by CD80 and interleukin-15. Nat Med.
2003;9(3):279-86.
67. Hollyman D, Stefanski J,
Przybylowski M, Bartido S,
Borquez-Ojeda O, Taylor C, et al. Manufacturing validation of
biologically functional T cells targeted to CD19
antigen for autologous adoptive cell therapy. J Immunother.
2009;32(2):169-80.
68. Porter DL, Levine BL, Kalos M,
Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic
lymphoid leukemia. N Engl J Med. 2011;365(8):725-33.
69. Kalos M, Levine BL, Porter DL,
Katz S, Grupp SA, Bagg
A, et al. T cells with chimeric antigen receptors have
potent antitumor effects and can establish memory
in patients with advanced leukemia. Sci Transl Med.
2011;3(95):95ra73.
70. Grupp SA, Kalos M, Barrett D,
Aplenc R, Porter DL,
Rheingold SR, et al. Chimeric antigen receptor-modified T cells for
acute lymphoid leukemia. N Engl J Med.
2013;368(16):1509-18.
71. Brentjens RJ, Davila ML, Riviere
I, Park J, Wang X,
Cowell LG, et al. CD19-targeted T cells rapidly induce
molecular remissions in adults with chemotherapy-refractory acute
lymphoblastic leukemia. Sci Transl Med.
2013;5(177):177ra38.
72. Maude SL, Laetsch TW, Buechner J,
Rives S, Boyer M,
Bittencourt H, et al. Tisagenlecleucel in Children and
Young Adults with B-Cell Lymphoblastic Leukemia. N
Engl J Med. 2018;378(5):439-48.
73. Neelapu SS, Tummala S, Kebriaei
P, Wierda W, Gutierrez C, Locke FL, et al. Chimeric antigen receptor
T-cell
therapy - assessment and management of toxicities. Nat
Rev Clin Oncol. 2018;15(1):47-62.
Recibido:
Noviembre 10, 2020
Aprobado: Noviembre 27, 2020
Correspondencia:
Beatriz Wills-Saní
willssab@mskcc.org