Resumen
Los primeros datos sobre el sarcoma aparecieron en escritos del año
1500 a.C., aunque no fue hasta el
descubrimiento del microscopio cuando se empezó a progresar en su
estudio y clasificación. El pilar de
su tratamiento ha sido y es la cirugía, que fue evolucionando desde una
escisión simple a tratamientos
radicales como la amputación o la resección amplia con márgenes,
reduciendo así el riesgo de recurrencia.
Paralelamente a la aceptación de la preservación de extremidad, como
mejor tratamiento quirúrgico fue
planteando la introducción del manejo multimodal, inicialmente con
radioterapia. Tanto la cirugía como
la radioterapia se han visto beneficiadas por los avances en las
técnicas de imagen, que han aportado un
mejor conocimiento de la anatomía. La quimioterapia se introdujo en el
estadio localizado con la finalidad de tratar las posibles
micrometástasis, y en el escenario metastásico. No obstante, los
tratamientos sistémicos tienen un recorrido histórico más corto y su
progresión ha sido menor. Profundizar en la biología
molecular y las diferentes sensibilidades según subtipos histológicos,
podrían aportar un nuevo abanico
de opciones terapéuticas.
Palabras clave: Sarcoma; historia; progreso; diagnóstico;
tratamiento.
SARCOMAS: CHANGES IN HISTORY
Abstract
The first data on sarcoma emerged in writings from 1500 BC, although
it was not until the discovery of the microscope that progress in its
study and classifi cation started. The mainstay of
its treatment has been and still is surgery, which has evolved from a
simple excision to radical
treatments, such as amputation or wide resection with margins,
decreasing the risk of recurrence.
Simultaneously with the acceptance of limb salvage, as the best
surgical treatment, the introduction of multimodal management was
considered, initially with radiotherapy. Both surgery and radiotherapy
have benefi ted from advances in imaging, which have provided a better
knowledge of
anatomy. Chemotherapy was introduced into the localized stage in order
to treat potential micrometastases, and into the metastatic scenario.
However, systemic treatments have a shorter historical trajectory and
their progress has been minor. Delve into molecular biology and the
diff erent
sensitivities according to histological subtypes, could provide a new
range of therapeutic options.
Keywords:
Sarcoma; history; progress; diagnosis; treatment

¹ Departamento de Oncología Médica, Sección Sarcomas,
Instituto Catalán de Oncología, ICO, Badalona, Cataluña, España
Introducción
El término sarcoma deriva del griego sarx que significa
carnoso, por lo que un
sarkoma
sería, literalmente, una
excrecencia carnosa. Esta palabra se hizo popular para
denominar a los tumores malignos de tejidos blandos
y huesos (1).
Factores de riesgo
Durante su historia, se han planteado distintos factores
de riesgo para su desarrollo. Uno de los más sugeridos
ha sido el traumatismo previo. El primero en plantearlo fue Everad
Home, en 1804, describiendo el caso de
un paciente con un hematoma, que posteriormente padeció un tumor
muscular (2). El caso fue denegado por
la compañía de seguros, ya que varios médicos desaprobaron la relación
causa - efecto entre el trauma y el
tumor posterior. Esta teoría fue revalorada en 1926 por
el director de patología del Hospital Memorial, James
Ewing (1866-1943), quien publicó un documento en
contra de distintos patólogos, que concluían que más
del 40% de los sarcomas eran de origen traumático (3).
A pesar de sus argumentos, la duda sobre la asociación
entre trauma y sarcoma persistió.
Debe destacarse, también, el descubrimiento de F.
Peyton Rous (1879-1970) en 1910, quien identificó
un virus causante de sarcomas en pollos, conocido
desde entonces como “el virus del sarcoma de Rous”.
Al aislar e inocular ese virus en animales sanos, el
tumor se reproducía en ellos (4). Por lograr el cultivo
y crecimiento del primer sarcoma, in vitro, Rous recibió el Premio
Nobel en 1966, 56 años después de su
hazaña.
Por último, el hecho de que los carcinógenos químicos
indujeran sarcomas en animales, expuso la cuestión de
si algún agente químico podría desarrollar un sarcoma
en humanos. Lamentablemente, la rareza del tumor
hizo difícil demostrar este nexo, aunque sí se probó el
aumento del riesgo de padecer sarcoma en población
expuesta a ácidos fenoxiacéticos o clorofenoles (5).
Estos tumores requieren de una visión mutidisciplinar,
por un equipo experto, desde su diagnóstico hasta su
tratamiento. Cada una de las especialidades implicadas en su
valoración, han sufrido una evolución a lo
largo de la historia. A continuación, se detallan los
cambios más destacables.
Evolución en el diagnóstico
Anatomía patológica
Los sarcomas son tumores infrecuentes, de origen mesenquimal, e
incluyen un amplio número de subtipos.
Esto supone un gran desafío diagnóstico, tanto para su
identificación como para su clasificación en un subgrupo histológico.
No obstante, fue hasta el siglo XVIII, en
el que los avances tecnológicos y médicos permitieron
progresos relevantes en su conocimiento. El desarrollo
del microscopio acromático, las técnicas de sección fina
de tejido y el reconocimiento de las diferentes capas
embrionarias, fueron contribuciones destacables (6). De
hecho, el descubrimiento del microscopio, en 1592, fue
crucial para la clasificación moderna de los sarcomas
(6). Esta clasificación histológica ha evolucionado durante los años,
siendo la última revisión de la Organización Mundial de la Salud (OMS)
la publicada en 2020,
en la que se especifican más de 80 subtipos.
Las primeras descripciones de sarcomas, y de cáncer
en general, se remiten al antiguo Egipto. El Papiro de
Smith, basado en los conocimientos en medicina y
cirugía del año 3000 a.C., ya contiene referencias sobre el cáncer (7).
Por su parte, el Papiro de Ebers, del
1500 a.C., menciona los tumores de tejidos blandos,
llamados “tumores grasos”. La recomendación, en ese
momento, era la de tratarlo con un bisturí, aunque, si
el tumor era grande y localizado en una extremidad, se
aconsejaba no llevar a cabo ningún procedimiento (8).
Claudio Galeno (160 d.C.), médico griego, también
escribió sobre cáncer y sarcomas, definiendo el cáncer como una
enfermedad sistémica, cuyo origen era
el exceso de bilis negra. Esta teoría había sido expuesta previamente
por Hipócrates, quien también había
descrito “tumores superficiales y profundos en brazos
y piernas de gente mayor” (9). La hipótesis de la bilis
negra se mantuvo firme hasta el siglo XVIII.
En 1632, el médico italiano Marco A. Severino (1580
- 1656), escribió el primer libro de patología quirúrgica. En él,
englobaba todas las lesiones inflamatorias
bajo el término de absceso y citaba específicamente a
los sarcomas, detallando un mixosarcoma de un modo
tan preciso que sería reconocible en la actualidad (10)
Giovanni B. Morgagni (1682-1771), considerado padre
de la neuropatología, dedicó un capítulo a los tumores
en su libro “los asientos y las causas de las enfermedades
investigadas por la anatomía”, exponiendo su anatomía
patológica, mediante el examen de autopsias (11). Uno
de sus casos, evoca un liposarcoma de la época presente. También
destacó hallazgos similares de otros autores, con tumores gigantes, que
eran, sin duda, sarcomas.
Morgagni planteó el diagnóstico del cáncer como enfermedad localizada,
en sus estadios tempranos, y describió
su diseminación mediante el sistema linfático. Su trabajo
fue más allá, diferenciando cánceres de gomas, estromas
y exostoses y fundando la anatomía patológica basada a
nivel del órgano. Otro médico que clasificó los tumores
fue Thomas Hodgkin (1798-1866). Los agrupó en “scirrhous”, actuales
carcinomas, “fungoid”, considerados
hoy día linfomas y sarcomas, y “melanosis”, para los
melanomas. En su documento explica perfectamente la
“enfermedad fungoide” con origen óseo, desarrollando
lesiones grandes de crecimiento rápido, entidad identificable como lo
que entendemos por sarcoma (12) .
En 1803, el cirujano inglés William Hey (1736-1819)
escribió un capítulo llamado “Fungus Haematodes”,
incluyendo diez casos de pacientes con tumores en
extremidades, fungoides y altamente vascularizados
(13). Siguiendo su estela, el cirujano escocés, Wardrop
(1782-1869), determinó que ese “Fungus Haematodes” era un tumor de
tejidos blandos. Después de huir
de Francia, cuando Napoleón perseguía a los residentes ingleses para
arrestarlos, adquirió sus conocimientos en Viena y escribió un libro
con varios casos de esta
entidad, añadiendo ilustraciones (14).
Al año siguiente, John Abernethy (1764-1831), también británico, hizo
una primera clasificación clínica y
anatómica de un conjunto de enfermedades a las que
llamó sarcomas, usando la vieja definición de “tumor
carnoso”. Dentro de este grupo incluía inflamaciones,
quistes, infecciones y aneurismas y los nombraba según su parecido a
los órganos (Tabla 1). Entre los tumores descritos por Abernethy,
también se identifican,
claramente, casos a los que actualmente llamaríamos
sarcomas (15).
Tabla 1. Primera clasificación histológica de los sarcomas
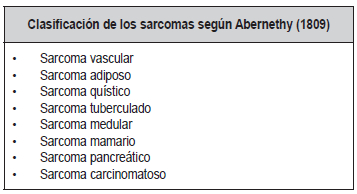
Hasta ese momento, nadie había categorizado los tumores según lo que
ahora conocemos por “sarcomas”,
sino que utilizaban el término para nombrar diferentes
entidades, entre las que se iban exponiendo casos de
sarcomas como tal. No obstante, muy poco después,
en 1805, el francés Alexis Boyer (1757-1833), cirujano
personal de Napoleón, citó entidades como la exóstosis o osteocondroma,
goma de hueso, espina ventosa
(que significa hueso corto expandido con aire) y osteosarcoma (16)
No fue sino hasta 1845, cuando Hermann Lebert
(1813-1878), médico con interés en anatomía, publicó
un atlas ilustrado en el que apareció, por primera vez,
el aspecto microscópico de un verdadero sarcoma de
tejidos blandos (17). A raíz de ese acontecimiento, se
empezó a progresar en el estudio de distintos tipos de
sarcomas, con Rudolph Virchow (1821-1902) y Theodor Schwann (1810-1882)
como impulsores. En 1860,
Virchow describió los sarcomas según sus estructuras
microscópicas, separándolos en 6 tipos: fibrosarcoma,
mixosarcoma, gliosarcoma, melanosarcoma, condrosarcoma y osteosarcoma
(18). El término angiosarcoma también fue introducido y un dermatólogo
húngaro, llamado Kaposi (1837-1902), expuso los primeros
casos de una enfermedad cutánea, nombrada, desde
ese momento, “Sarcoma de Kaposi” (19).
Samuel W. Gross (1805-1884), en 1879, publicó el
primer artículo dando una visión global del sarcoma,
detallando 165 casos. En él, exponía su histología, clínica,
diagnóstico, pronóstico y tratamiento. Su agrupación de sarcomas óseos
se basó en la localización,
central o periostal, y el tipo celular, células gigantes,
en huso o redondas. También explicó la tendencia a
la diseminación hematógena, hacia los pulmones, con
poca afectación linfática (20).
Durante un tiempo, hubo diversidad de opiniones en
cuanto a la malignidad del sarcoma. Frank B. Mallory
(1862-1941) fue el primero en definir, histológicamente, diferentes
tipos de sarcomas, utilizando tinciones
(21). Gracias a ellas, Max Borst (1869-1946) estableció, de forma
definitiva, que los sarcomas son tumores
mesenquimales malignos y nombrar nuevas entidades,
como en hemangioendotelioma, linfangioendotelioma y peritelioma
(posteriormente llamado hemangiopericitoma) (22).
En referencia al liposarcoma, en concreto, después de
una descripción hecha por Virchow, en 1865, hubo
laxitud en llamar liposarcoma a cualquier neoplasia
con origen en el tejido adiposo. En 1925 Caldwell y
Zinninger propusieron dividir el término liposarcoma en tres entidades:
Lipomiosarcomas, lipomas con
áreas de degeneración sarcomatoide (normalmente
fibrosarcomas con células en huso) y verdaderos liposarcomas (23).
Progresivamente, se fueron añadiendo nuevos subtipos
de sarcomas y, en los años 40, Arthur P. Stout (1885-
1967) publicó una serie de escritos sobre distintas lesiones
mesenquimales, como los mixomas, fibrosarcomas y leiomiomas,
incorporando el schwannoma
maligno, histiocitoma fibroso maligno y mesenquimoma. Igualmente,
clasificó los liposarcomas en 4 tipos:
bien diferenciado de tipo mixoide, pobremente diferenciado de tipo
mixoide, adenoide o de células redondas y mixoide (24). El mismo fue
quien precisó que el
sarcoma tiene su origen en las células mesenquimales
que conforman las estructuras de soporte, pudiendo
contener más o menos tejidos diferenciados, sin predominancia de
ninguno (25). En 1953 definió una
nueva clasificación. La introducción del microscopio
electrónico, hizo replantear esta última clasificación y
descubrir nuevas entidades como el rabdomiosarcoma
alveolar, el sarcoma epitelioide y el dermatofibrosarcoma
metastatizante (26).
El primer libro con la clasificación histogénetica de los
tumores de tejidos blandos, llevada a cabo por la OMS,
se imprimió en 1969 (27).
En relación con la estadificación del sarcoma, hay que
destacar la evolución distinta que ha sufrido este tipo
tumoral, en comparación con el resto de cánceres.
El sistema TNM (del inglés: tumor, lymph node, metastases) fue
desarrollado por Pierre Denoix, en 1960,
bajo la autoridad de la “International Union Against
Cancer” (UICC), y revisado por el “American Joint
Committee on Cancer” (AJCC). Su conclusión fue que
el sistema internacional TNM era inadecuado para los sarcomas, por lo
que, tras años de trabajo, en 1977 se
publicó un sistema TNM propio para ellos, añadiendo
el grado histológico (28) (Tabla 2). Aunque esta nueva clasificación
duró poco, decidiendo tener en cuenta,
no sólo el grado y el tamaño tumoral, sino también la
profundidad del tumor (superficial o profundo). Así, en
1979, se estableció el sistema de estadificación conocido
actualmente como “Memorial Sloan-Kettering”, publicándose en el libro
“Pathology of Soft Tissue Tumors”
(29). Esta clasificación se aceptó en 1990 por la UICC,
AJCC y diferentes sociedades oncológicas.
Diagnóstico por la imagen
Los rayos-X, descubiertos por Wilhelm C. Roentgen
(1845-1923) en 1895, fueron introducidos rápidamente
en el campo de la medicina como herramienta diagnóstica. El cirujano
Ernest A. Codman (1869-1940),
entusiasta del estudio de los tumores óseos, publicó
“El uso de los rayos-X en el diagnóstico de enfermedades óseas” (30) y
contribuyó en la fundación de la escuela americana de cirujanos.
Animado por el interés
suscitado en muchos cirujanos, y gracias a la donación
de la familia de un paciente, en 1921, junto a Joseph
C. Bloodgood (1867-1935) y James Ewing, inició un
registro americano de sarcomas. El amplio número de
casos recopilados, tuvo un gran valor para el aprendizaje de su
diagnóstico y tratamiento, destacando la
importancia de una correlación clínico - radiológica e
histológica y ayudando a estandarizar el manejo de los
pacientes en cuanto a terapias y seguimientos (1).
La resonancia magnética (RM), una de las pruebas
más valiosa en los sarcomas, está basada en el descubrimiento del Peter
Mansfield y Paul C. Lauterbur,
galardonado con un premio Nobel y fue aprobada en
1984 por la “Food and Drug Administration” (FDA).
Esta técnica no invasiva supuso un avance, sin precedentes, en el
conocimiento de la anatomía, mejorando
la planificación quirúrgica y el delineado tumoral, y
del edema adyacente, en la irradiación (31).
Otras técnicas, como la angiografía, la tomografía axial
computarizada (TAC) y la introducción del tecnecio
marcado, también fueron decisivas para el progreso
en las intervenciones quirúrgicas. Así, permitieron a
los cirujanos un mejor conocimiento de la anatomía
de las lesiones y su relación con el tejido circundante, ofreciendo una
mayor seguridad para realizar procedimientos más conservadores. Además,
también contribuyeron a cambiar el método de diagnóstico histológico
prequirúrgico, subtituyendo a la biopsia abierta,
llevada a cabo bajo anestesia, por la biopsia cerrada
con aguja, reduciendo complicaciones (31).
Tabla 2. Primera estadificación TNM de los sarcomas (1977)
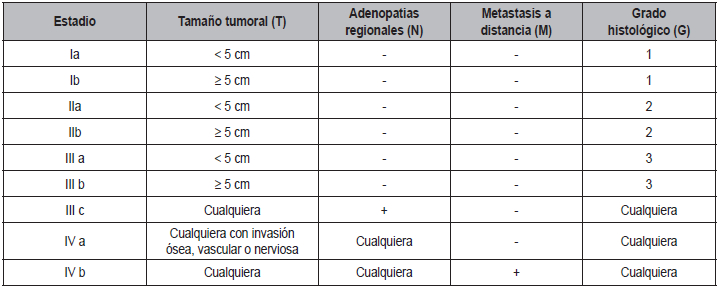
El último adelanto en imagen, con impacto en el sarcoma, fue el
desarrollo de la tomografía por emisión
de positrones (PET). Gracias a la lectura de la actividad metabólica
tumoral, permite detectar recidivas
precoces y valorar anticipadamente la respuesta a un
tratamiento. Así mismo, las TAC y RM con neuronavegador o a tiempo
real, han ayudado a guiar tanto las
cirugías, como las técnicas termoablativas (criopreservación y
radiofrecuencia) (31).
Evolución en el tratamiento
Cirugía
Aunque a hoy pueda parecer obvio que el tratamiento
del sarcoma debe implicar una cirugía, no siempre fue
así. Durante siglos, Celsio (25 a. C. - 50 d. C.) y Galen (131 -200 d.
C.) (médicos griegos), y muchos de sus
compañeros, desaconsejaban la cirugía de los tumores,
incluidos los sarcomas, cuando tenían forma irregular,
color pálido, úlcera, insensibilidad o no podían ser movilizados con
los dedos. En la misma dirección Teodoro
de Salerno (1205-1296) señalaba que los tumores lipomatosos, oscuros y
de consistencia firme, eran cancerosos, y que la cirugía no era una
maniobra adecuada
para ellos. Finalmente, Guy de Chauliac (1300-1368),
cirujano francés y médico personal de tres Papas, aportó una nueva
valoración, recomendando una cirugía
amplia para los tumores en fase inicial, cuando la lesión era pequeña y
superficial (26). Hacia el final del
1700, se aceptó que cualquier tipo de cáncer, incluido
el sarcoma, debía tratarse con cirugía. A partir de ese
momento, el manejo del sarcoma siempre ha implicado una intervención
quirúrgica, más o menos extensa,
como tratamiento principal (26). En este sentido, fue
el cirujano inglés John Hunter (1728-1793), quien propuso que toda
lesión cancerígena se escindiera junto al
tejido circundante, siendo el primero en aconsejar una
cirugía con márgenes (26).
Ewing también añadió un punto interesante al procedimiento quirúrgico,
determinando la necesidad del
diagnóstico prequirúrgico sistemático en todos los tumores de tejidos
blandos y óseos, mediante citología
por aspiración (26).
En cuanto a la técnica quirúrgica propiamente dicha,
durante las décadas de 1930 y 40, el abordaje de los
sarcomas se basó en la cirugía exclusiva, con escisión
simple. No obstante, los cirujanos de la época, pronto se percataron de
que ese enfoque era subóptimo
en los sarcomas de extremidades, relacionándose con
una tasa de recurrencia mayor al 50% (60-95%), siendo más elevada en
algunos subtipos, como el sarcoma
sinovial. También se determinó la asociación entre la
recurrencia y el riesgo de metástasis y de muerte relacionada con el
sarcoma (32, 33). Por el contrario, en
los casos de resección amplia o amputación, las recurrencias eran
menores (25-30%)(32).
Con el fin de indagar más sobre el mejor abordaje quirúrgico, Albert B.
Ferguson (1895-1976) decidió revisar 258 casos de pacientes con sarcoma
óseo, tratados
mediante amputación, extraídos del registro americano previamente
mencionado. Su conclusión fue que,
aunque la amputación precoz era un tratamiento comúnmente utilizado, no
sólo no era el mejor sino que
provocaba la muerte del paciente (34). Llegó a esa deducción
objetivando que los pacientes con amputación
precoz (entre los 6 meses posteriores al inicio de los
síntomas) tenían una supervivencia del 8%, mientras
que los amputados a partir de ese momento tenían
una supervivencia del 28%. Sus recomendaciones fueron: evitar la
amputación precoz; irradiar, extirpar e
implantar injertos óseos previamente; extirpar si había una recurrencia
evidente y finalmente amputar. Es indiscutible que el retraso en la
cirugía implica un sesgo, a favor de tumores de lento crecimiento y
menos
agresivos y en contra de lesiones de rápida evolución
con metástasis tempranas (pacientes no tributarios a
un abordaje local transcurridos 6 meses). Aún así, el
protocolo de Ferguson fue seguido durante años por
múltiples cirujanos, lo que llevó a retrasar la aceptación de que una
cirugía radical y precoz es el mejor
tratamiento quirúrgico en estos pacientes.
A pesar de la baja supervivencia, incluso tras una amputación, se
empezó a plantear el papel de una cirugía
conservadora de miembro, con resección compartimental. Dallas B.
Phemister (1882-1951) fue uno de
los pioneros en introducir el concepto de cirugía de
preservación de extremidad, en los años 50, poco antes de su muerte
prematura a causa de una apendicitis
(35). Siguiendo esa misma idea, en los años 60 se publicaron diferentes
textos exponiendo que, aunque la
escisión simple era eficaz en la eliminación del tumor
palpable, su baja supervivencia libre de enfermedad local se debía a la
persistencia de células cancerígenas
en los márgenes de la cicatriz quirúrgica (36). De ese
modo, se sugirió la ampliación del área de resección,
con el fin de eliminar la enfermedad microscópica, no
incluida en un campo de escisión simple (32). La sustitución de la
escisión simple por una resección radical,
incluyendo el recorrido de la biopsia y márgenes amplios, redujo
dramáticamente la tasa de recurrencias,
de un 70-95% a un 10-30% (37).
También en la década de los 60, Frank F. Parrish (1911-
1979) destacó las ventajas de la escisión en bloque frente
al curetaje, para los tumores malignos óseos, y describió
el uso de injerto de hueso largo para la reconstrucción
de los déficits posquirúrgicos (38). En el campo de los
injertos, se debe destacar a William Enneking (1926-
2014), quien introdujo la técnica de resección-artrodesis
para la preservación de extremidad, utilizando injertos
con raíz intramedular; a Henry J. Mankin (1928-2018),
quien reconstruyó con aloinjertos; y a Ralph C. Marcove (1929-2001), el
primero en usar implantes metálicos
(endoprótesis) para el reemplazo de huesos como el fémur (31).
Posteriormente, las mejorías en este campo
se focalizaron en el uso de diferentes tipos de prótesis e
injertos, valorando el impacto funcional y el riesgo de
infección quirúrgica.
Así pues, conforme la preservación de extremidad se
fue convirtiendo en una técnica aceptada universalmente, los cirujanos
empezaron a plantearse el impacto de la cirugía en la calidad de vida
de los pacientes.
Paul Sugarbaker fue el primero en usar diferentes escalas para
evaluarla. No obstante, su análisis, con pocos
pacientes incluidos, no pudo demostrar un beneficio
significativo en la conservación de extremidad seguida
de radioterapia frente a la amputación (39). En 1993,
Enneking publicó un sistema de valoración del resultado funcional
esperado en pacientes tras cirugía reconstructiva, método que se ha ido
desarrollando posteriormente (40). Finalmente, en 2007 se evidenció
que,
la mayoría de pacientes afectos de sarcoma, eran candidatos a una
cirugía con preservación de extremidad,
obteniendo estos el doble de los resultados funcionales
satisfactorios en comparación con la amputación (41).
En cuanto a la resección de las metástasis, cabe mencionar a Judson
McNamara, el pionero en introducir
el potencial de curabilidad con la escisión de las metástasis
pulmonares. En concreto, describió la extirpación
de una metástasis pulmonar solitaria en un paciente
con osteosarcoma, a finales de los 60 (42).
De forma paralela a los avances quirúrgicos, se fue
investigando si añadir tratamiento pre o posquirúrgico, ya fuera con
quimio o radioterapia, podría
mejorar la supervivencia libre de progresión (SLP)
local o supervivencia global (SG). La combinación
apropiada de estas terapias y la cirugía fueron los
principales temas de estudio en el área del sarcoma,
en el siglo XX (6).
Radioterapia
En 1898, el matrimonio Curie, tras años investigando la radioactividad,
descubrió el radio y, cinco años
después, se empezó a usar como tratamiento contra el
cáncer. El empleo de este elemento interesó a James
Douglas, un filántropo ingeniero de minas, quien fundó, junto a James
Ewing, el “Memorial Hospital for
Cancer and Allied diseases” y estableció un departamento de radio. De
este modo, sentaron las bases para
el desarrollo de la radioterapia, como tratamiento del
cáncer, en los Estados Unidos. En 1917, un tercio del
suministro mundial de radio se hallaba en su centro
(1). En 1920, Ewing, identificó el “endotelioma difuso
del hueso”, llamado “Tumor de Ewing”, con especial
sensibilidad a la radio y quimioterapia, convirtiéndose
en un entusiasta de la irradiación, también en liposarcomas y sarcomas
con componente mixoide (43).
No obstante, durante años se catalogó al sarcoma
como un tumor radiorresistente, proponiendo la irradiación únicamente
como tratamiento paliativo, en
casos avanzados e irresecables (44). El primer caso de
un paciente con sarcoma, tratado con radioterapia,
con intención radical, se describió en el 1935. Era
un paciente afecto de un fibrosarcoma recurrente, escapular, tratado
con rayos-X profundos. Al cabo de
10 años, el paciente seguía libre de recurrencia (45).
Poco a poco, se fue recopilando información de casos
tratados y se probó que los sarcomas podían responder a esta terapia.
De modo que, con el tiempo, pasó
a considerarse una opción como tratamiento radical,
sobre todo en lesiones pequeñas, e incluso plantearse su papel
posoperatorio (36, 44). La combinación
de la cirugía y radioterapia fue ganando interés, conforme se fue
poniendo en duda la amputación como
mejor tratamiento radical y planteando tratamientos
más conservadores.
En el escenario prequirúrgico, Edward L. Atkinson
(1881-1929), en 1963, fue el primero en describir el
uso de radioterapia en 15 pacientes con sarcoma. Su
objetivo era el de encapsular el tumor y beneficiarse
del propio efecto antitumoral. La dosis utilizada fue
de 4.500 rads en 4 - 5 semanas, aplicándose entre 4 y
6 semanas antes de la resección. Con una mediana de
seguimiento de 3 años, se diagnosticó una recurrencia
local y ninguna progresión a distancia (33).
En los años 70, se publicaron resultados de casos tratados con
preservación de extremidad y radioterapia
adyuvante. Algunos autores aconsejaban la resección
simple del tumor visible, seguida de radioterapia (6.500
rads en 6,5 semanas), y, para el rabdomiosarcoma o
sarcoma sinovial, quimio-radioterapia concomitante
y posterior mantenimiento con quimioterapia. Para
sarcomas de rápido crecimiento, primarios o recurrentes, se sugería
radioterapia inmediata y valoración
quirúrgica a posteriori. (37) A finales de esa década,
la recomendación era más específica. Así, en estadios
iniciales se indicaba una cirugía radical con márgenes
amplios, añadiendo radioterapia adyuvante en caso
de márgenes inadecuados o neoadyuvante, si se podía
anticipar una cirugía no radical. En los estadios IIA
- IVA, la radioterapia neoadyuvante era un estándar,
a dosis de 5.000 - 5.600 rads, pudiendo agregar una
impresión de 1.000 - 1.500 rads intra o posquirúrgica (36). Hasta ese
momento, los grupos incluidos en
los estudios eran heterogéneos, pero se fueron compilando datos sobre
la histología, el grado tumoral, la
localización y el tamaño. Se empezaba a contemplar
que 5 cm podría ser el tamaño límite que dividía dos
grupos con excelente o buen resultado al tratamiento,
así como que el grado tumoral se relacionaba con la
recurrencia. Estos factores parecían no tanto un factor
predictivo sino pronóstico (46).
Finalmente, ya en los años 80, la radioterapia adyuvante se convirtió
en práctica habitual, gracias a las
conclusiones de un estudio prospectivo randomizado.
Este ensayo incluyó 43 pacientes y demostró que el
grupo que recibía cirugía conservadora con radioterapia posterior (27
pacientes), obtenía resultados equivalentes a la amputación (16
pacientes) en SG, supervivencia libre de enfermedad y calidad de vida
(47) .
Hasta ese momento, no había datos comparativos entre el tratamiento
neoadyuvante o adyuvante. Si bien,
el tratamiento preoperatorio tenía beneficios obvios,
como: el menor campo de irradiación y menor daño
de tejido sano, por una mejor delineación, y la disminución del riesgo
de diseminación intraquirúrgica,
por reducción y encapsulación del tumor. En 2002,
se llevó a cabo un estudio randomizado, en pacientes
con sarcoma, que recibían radioterapia pre o postquirúrgica. Este
demostró un discreto beneficio en SG
en el grupo de tratamiento neoadyuvante. Las tasas
de recurrencia local o a distancia y la SLP fueron
iguales entre los grupos. Aunque las complicaciones
en la cicatrización fueron mayores con la administración prequirúrgica,
no se objetivaron secuelas a largo
plazo (48).
En la actualidad, se consideran factores de alto riesgo
el tamaño tumoral (> 5 cm), profundidad, alto grado
(grado 2-3) y localización en extremidades o tronco
(49). En estos casos está indicada la radioterapia, teniendo en cuenta
que, si bien sigue habiendo controversia, el tratamiento preoperatorio
es el más aceptado.
Las investigaciones basadas en la técnica de radioterapia han permitido
evolucionar desde la visión bidimensional a la tridimensional e
implementar nuevos
métodos, con el fin de reducir la toxicidad y mejorar
la calidad de vida, manteniendo la eficacia. La IMRT
(radioterapia de intensidad modulada) y la IGRT (radioterapia guiada
por imagen) permiten una mayor
precisión, con la administración de una dosis alta en
la región diana y una menor irradiación de la zona circundante (50).
Los últimos estudios proponen la combinación de la radiación con
distintos fármacos, con la
finalidad de aumentar la radiosensibilidad.
Tratamiento sistémico
Curiosamente, una de las primeras terapias documentadas para el
tratamiento de los sarcomas, fue la inmunoterapia. En 1891, William B.
Coley (1862-1936)
observó cómo un paciente con un sarcoma recurrente
de células redondas pequeñas irresecable, sufría una
regresión tumoral al padecer erisipela. A raíz de esta
experiencia, Coley empezó a utilizar distintas toxinas
bacterianas para tratar sarcomas de tejidos blandos y
hueso inoperables (51). A pesar de sus buenos resultados en más de
1.000 pacientes, Coley fue muy criticado y muchos de sus compañeros
pusieron en duda sus
resultados. Esto, junto a la aparición de nuevos tratamientos, como la
radio y quimioterapia, hizo que las
“toxinas de Coley” quedaran en el olvido.
En los últimos años, se ha retomado el interés por la
inmunoterapia en el tratamiento del cáncer, y del sarcoma en concreto.
Algunos subtipos determinados,
como el sarcoma alveolar de partes blandas o sarcoma pleomórfico
indiferenciado han mostrado especial
sensibilidad.
La historia del resto de agentes sistémicos, utilizados
en estos tumores, se encuentra en un escenario mucho
más actual, ya que empezaron a utilizarse hace únicamente unos 50 años (
Figura 1).
En los tumores óseos, como el osteosarcoma de extremidad y el sarcoma
de Ewing, la quimioterapia ha tenido
un impacto destacable. La introducción del tratamiento
estándar en el osteosarcoma (doxorrubicina, cisplatino,
metotrexate a altas dosis e ifosfamida) y el del sarcoma
de Ewing (vincristina, doxorrubicina, ciclofosfamida,
ifosfamida y etopósido) aumentó la tasa de curación del
20% (con cirugía sola) a cerca del 60%. A partir de aquí,
el único progreso destacable fue la adición de MTP-PE
(Muramil Tripéptido Fosfatidiletanolamina Encapsulado en Liposomas) en
osteosarcomas, con mejoría de la
supervivencia libre de evento (52).
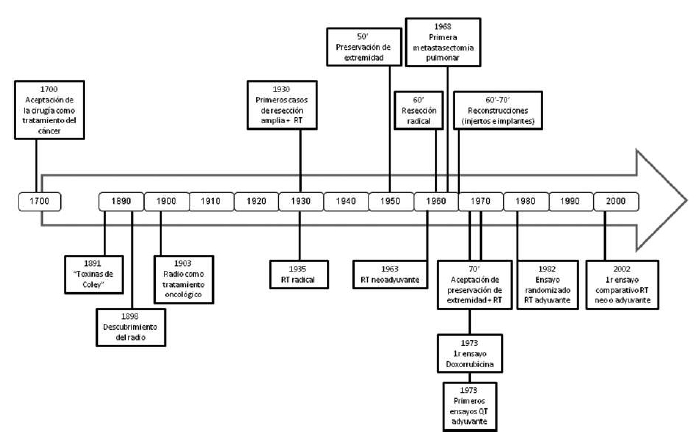
Figura 1. Hitos destacables en la evolución del tratamiento del
sarcoma. RT: radioterapia.
Por otro lado, aunque en los pacientes con sarcoma
la cirugía fue, y sigue siendo, el pilar fundamental
del tratamiento, un elevado número de ellos recaían,
muchas veces en forma de lesión irresecable o metastásica, acortando
drásticamente supervivencia. Este
hecho, hizo plantearse si añadir quimioterapia, en estadio localizado,
podría aumentar la SG. No obstante,
la historia de la quimioterapia en el sarcoma de tejidos
blandos no ha representado un adelanto tan señalable,
muy posiblemente por la menor quimiosensibilidad de
este grupo en general. La gran heterogeneidad de los
sarcomas hace difícil su representatividad en los ensayos clínicos y
los estudios específicos por subgrupos
constan de pocos pacientes. Esto lleva a que la valoración de la
eficacia de los fármacos sea compleja (52).
Doxorrubicina se evaluó por primera vez en 1973, en
un ensayo fase II en el que se incluyeron pacientes con
múltiples tumores. Los sarcomas mostraron especial
sensibilidad (21 remisiones en 64 pacientes) (53).
Pese a que el objetivo de este documento no es la revisión sistemática
de todos los ensayos clínicos, cabe
destacar algunos resultados generales que hicieron
cambiar la práctica habitual en el sarcoma. En contexto adyuvante, se
publicaron distintos estudios randomizados, comparando cirugía sola,
frente a diversos
esquemas quimioterápicos basados en antraciclinas
(54). A pesar de que las poblaciones eran muy heterogéneas, dos de los
ensayos demostraron un aumento
en SG en la rama de quimioterapia, mientras que nueve no encontraron
diferencias significativas. Distintos
metaanálisis también intentaron esclarecer este papel
de la quimioterapia en la adyuvancia. En 1997, se publicó uno con datos
de más de 1.500 pacientes, mostrando un beneficio significativo en
relación al intervalo libre de recurrencia local y a distancia, aunque
el
incremento en SG no fue significativo (55). La mayor
evidencia fue para los sarcomas localizados en extremidades (HR 0,80;
p=0,029). En 2008, una actualización del metaanálisis con casi 2.000
casos, confirmó
la mejoría en intervalo libre de recurrencia para los
pacientes a los que se les administraba adyuvancia y,
además, ratificó un aumento en SG en los que recibían
la combinación de doxorrubicina - ifosfamida (56).
Actualmente se considera que el tratamiento adyuvante con ifosfamida y
doxorrubicina debe proponerse a
pacientes con sarcomas de extremidades, de alto riesgo (localización
profunda, > 5 cm y alto grado), a pesar de que no hay datos
definitivos de beneficio en SG.
La administración de manera neoadyuvante ofrece la
ventaja de probar la quimiosensibilidad y poder iniciar
el tratamiento de la enfermedad micrometastásica de
forma inmediata (49).
En el escenario de enfermedad metastásica, doxorrubicina sigue siendo
el tratamiento más activo, por lo
que se usa como tratamiento de primera línea. La cardiotoxicidad es su
principal toxicidad, por lo que, durante un tiempo se buscaron
antraciclinas alternativas.
Epirubicina, a dosis equimolar, no aportó beneficio en
toxicidad ni en actividad (57). Sí supuso una mejoría el
descubrimiento de la doxorrubicina liposomal, con eficacia similar y
menor toxicidad cardíaca, si bien estos
datos no se han confirmado en un estudio fase III (58).
Ifosfamida es el segundo agente más utilizado en sarcomas, aunque
durante años su uso fue limitado, debido a su asociación con la
cistitis hemorrágica, hasta
la introducción del mesna en 1979. En distintos ensayos, la dosis
estándar de 9 g/m2 no obtuvo ventajas al
compararla con doxorrubicina, por lo que actualmente
los dos son considerados los agentes más eficaces en
primera línea. La combinación de ambos, demostró
mayor tasa de respuestas (por encima del 50%), sin
impacto en SG, por lo que pasó a ser un tratamiento a
considerar en caso de intención de resecabilidad, en un
paciente apto para tolerar la biterapia (59) .
En 1970 se escribió, por primera vez, sobre el uso de
dacarbacina en pacientes con sarcoma, aportando
unas tasas de respuesta del 17% (60). En la actualidad,
se prefiere su uso en combinación con gemcitabina o
doxorrubicina, puesto que la SLP en monoterapia es de
solo 2 meses (61). Por su lado, la gemcitabina en monoterapia y en
combinación con docetaxel también se
ha ensayado para el tratamiento de estos tumores (62).
Los últimos citotóxicos descubiertos para el tratamiento del sarcoma,
trabectedina y eribulina, tienen origen
marino (un tunicado y una esponja, respectivamente).
Ambos demostraron superioridad frente a dacarbacina en pacientes con
leiomiosarcomas y liposarcomas
metastásicos pretratados. Trabectedina consiguió un
aumento en SLP (4,2 m vs 1,5 m) mientras que eribulina demostró mejorar
la SG (13,5 m vs 11,5 m), con
un aumento de 7 meses en los liposarcomas (63, 64).
En referencia a los tratamientos diana, pazopanib ha
sido el único en demostrar un beneficio en SLP (4,6 m
vs 1,6 m), en un ensayo fase III frente a placebo, incluyendo sarcomas
no adipocíticos (65).
En los últimos años, el avance en el tratamiento sistémico de los
sarcomas en general ha sido escaso, destacando únicamente tratamientos
en algunos sarcomas
específicos. Precisamente, las últimas investigaciones
se centran en el estudio de fármacos para determinados subtipos y en
profundizar en el conocimiento de su
biología molecular.
Conclusión
Los sarcomas conforman un grupo de múltiples subtipos histológicos, que
se han ido describiendo durante
siglos, hasta llegar a su clasificación actual. El mejor
conocimiento de este tumor ha venido determinado
por los avances radiológicos y patológicos junto con su interrelación
con la clínica. En los últimos 100 años, se
han producido progresos destacables en relación a su
tratamiento, sobre todo referente al manejo multidisciplinar. Aún así,
sigue siendo un tumor con mucho por
descubrir y con pocas opciones terapéuticas en estadio
avanzado. Aunque esto puede parecer desconsolador,
también forma parte del encanto de este tipo tumoral,
para el cual quedan múltiples puertas abiertas a la investigación.
Referencias
1. Peltier LF. Historical note on
bone
and soft tissue sarcoma. J Surg Oncol. 1985;30(4):201-5.
2. Home E. A short tract on the
formation of tumours. London: Longman, Hurst Rees Orme Brown &
Green; 1830.
3. Ewing_J. Relation of trauma to
malignant tumors. Amer
J Surg. 1926;40:30-6.
4. Rous P. A Transmissible Avian
Neoplasm. (Sarcoma of
the Common Fowl.). J Exp Med. 1910;12(5):696-705.
5. Hardell L, Sandstrom A.
Case-control study: soft-tissue
sarcomas and exposure to phenoxyacetic acids or chlorophenols. Br J
Cancer. 1979;39(6):711-7.
6. Sears HF. Soft tissue sarcoma: a
historical overview. Semin Oncol. 1981;8(2):129-32.
7. Breasted JH, New-York Historical
Society. Library. The
Edwin Smith surgical papyrus. Chicago, Ill.,: The University of Chicago
Press; 1930.
8. Ebbell_B. The Papyrus Ebers: the
greatest Egyptian
medical document. Copenhagen: Levinand Munksgaard; 1937.
9. Long ER. A history of pathology.
Baltimore: Williams and
Wilkins Co.; 1928.
10. Severinus MA. De abscessuum
recondita natura.
Naples, Beltranus; 1632.
11. Morgagni GB. The Seats and Causes
of Diseases investigated by Anatomy .Translated by Benjamin Alexander.
London: Millen&Cadell; 1769.
12. Hodgkin. On the Anatomical
Characters of some Adventitious Structures. Med Chir Trans. 1829;15(Pt
2):265-
448.
13. Hey WMRCS. Practical Observations
in Surgery, illustrated with Cases. London: Candell and Davis; 1803.
14. Wardrop J. Observations on fungus
haematodes or soft
cancer. Ramsay&Company, editor. Edinburgh; 1809.
15. Abernethy JMRCS. Tumor
classification in Surgical Observations. London: Hurst Rees Orme &
Brown Longman; 1804.
16. BoyerA. Traite des Maladies
Chirurgicales et des Operationes pi Leu Conviennent. Paris:Migneret;
1814.
17. LebertH. Physiologie
pathologique. Paris: JB Bailliere;
1845.
18. VirchowR. Die Krankhanften
Geschwuelste. Berlin;
1863.
19. KaposiM. Idiopathisches multiples
pigment-sarkom der
Haut. Arch Dermatol Syphil. 1872;4:265-73.
20. Gross SW. Sarcoma of the long
bones: Based upon a
study of 165 cases. Am J Med Sci 1879;78:17-57.
21. Mallory FB, WrightJH.
Pathological Tecnique. Philadelphia: WB Sanders; 1897.
22. BorstM. Die Lehre von den
Geschwulsten, mit einem
mikroskopischen Atlas. Wiesbadden: JF Bergmann;
1902.
23. Caldwell JA. Z. Extradural
liposarcoma of spinal canal;
clinical and pathological report. Surg Gynec and Obst.
1925;40:476-80.
24. Stout AP. Liposarcoma-the
Malignant Tumor of Lipoblasts. Ann Surg. 1944;119(1):86-107.
25. Stout AP. Tumors of the
musculoskeletal system in the
Altlas of Tumor Pathology. Washington DC: Armed Forces Institute of
Pathology; 1953.
26. Hajdu SI. Soft tissue sarcomas.
Cancer.
2007;109(9):1697-704.
27. Enzinger FM, Lattes R, Torloni H.
Histological typing of
soft tissue tumours: Geneva, World Health Organization;
1969.
28. Russell WO, Cohen J, Enzinger F,
Hajdu SI, Heise H,
Martin RG et al. A clinical and pathological staging system for soft
tissue sarcomas. Cancer. 1977;40(4):1562-
70.
29. Hajdu SI. Pathology of soft
tissue tumors. Philadelphia:
Lea and Febiger.London: Kimpton; 1979.
30. CodmanEA. The X-ray in surgery.
En: WW K, editor.
Surgery: its principles and practice. Philadelphia: WB
SaundersCo; 1919.
31. Henshaw RM. Surgical advances in
bone and soft tissue
sarcoma: 50 years of progress. Am Soc Clin Oncol Educ
Book. 2014.
32. Cantin J, McNeer GP, Chu FC,
Booher RJ. The problem
of local recurrence after treatment of soft tissue sarcoma. Ann Surg.
1968;168(1):47-53.
33. Atkinson L, Garvan JM, Newton NC.
Behavior and Management of Soft Connective Tissue Sarcomas. Cancer.
1963;16:1552-62.
34. FergusonAB. Treatment of
osteogenic sarcoma. J Bone
Joint Surg. 1940;22:92-100.
35. Phemister DB. Local resection of
malignant tumors of
bone. AMA Arch Surg. 1951;63(6):715-7.
36. Suit HD. Sarcoma of soft tissue.
CA Cancer J Clin.
1978;28(5):284-95.
37. Suit HD, Russell WO, Martin RG.
Management of patients with sarcoma of soft tissue in an extremity.
Cancer.
1973;31(5):1247-55.
38. Parrish FF. Treatment of bone
tumors by total excision
and replacement with massive autologous and homologous grafts. 1966.
Clin Orthop Relat Res. 2000(373):3-
10.
39. Sugarbaker PH, Barofsky I,
Rosenberg SA, Gianola FJ.
Quality of life assessment of patients in extremity sarcoma clinical
trials. Surgery. 1982;91(1):17-23.
40. Enneking WF, Dunham W, Gebhardt
MC, Malawar M,
Pritchard DJ. A system for the functional evaluation of
reconstructive procedures after surgical treatment of tumors of the
musculoskeletal system. Clin Orthop Relat
Res. 1993(286):241-6.
41. Enneking WF. History of
orthopedic oncology in the United States: progress from the past,
prospects for the future. Cancer Treat Res. 2009;152:529-71.
42. McNamara JJ, Paulson DL, Kingsley
WB, Urshel HC
Jr. Pulmonary resection for metastatic osteosarcoma.
JAMA. 1968;205(7):535-6.
43. Ewing J. Diffuse endothelioma of
bone. Proc NY Pathol
Soc. 1921;21:17-24.
44. Del Regato JA. Radiotherapy of
soft-tissue sarcomas.
JAMA. 1963;185:216-8.
45. LeucutiaT. Radiotherapy of
sarcomas of soft parts. Radiology. 1935;25:403-15.
46. Suit HD, Russell WO, Martin RG.
Sarcoma of soft tissue:
clinical and histopathologic parameters and response to
treatment. Cancer. 1975;35(5):1478-83.
47. Rosenberg SA, Tepper J, Glatstein
E, Costa J, Baker A,
Brennan M et al. The treatment of soft-tissue sarcomas
of the extremities: prospective randomized evaluations
of (1) limb-sparing surgery plus radiation therapy compared with
amputation and (2) the role of adjuvant chemotherapy. Ann Surg.
1982;196(3):305-15.
48. O'Sullivan B, Davis AM, Turcotte
R, Bell R, Catton C,
Chabot P et al. Preoperative versus postoperative radiotherapy in
soft-tissue sarcoma of the limbs: a randomised trial. Lancet.
2002;359(9325):2235-41.
49. Casali PG, Abecassis N, Aro HT,
Bauer S, Biagini R, Bielack S et al. Soft tissue and visceral sarcomas:
ESMOEURACAN Clinical Practice Guidelines for diagnosis,
treatment and follow-up. Ann Oncol. 2018;29(Suppl 4):
iv51-iv67.
50. El-Bared N, Wong P, Wang D. Soft
tissue sarcoma and
radiation therapy advances, impact on toxicity. Curr
Treat Options Oncol. 2015;16(5):19.
51. ColeyWB. The Treatment of
Malignant, Inoperable Tumors with the Mixed Toxins of Erysipelas and
Bacillus
Prodigiosus. Brussels: Weissenbruch; 1914.
52. Patel SR. Fifty years of advances
in sarcoma treatment:
moving the needle from conventional chemotherapy
to targeted therapy. Am Soc Clin Oncol Educ Book.
2014:259-62.
53. O'Bryan RM, Luce JK, Talley RW,
Gottlieb JA, Baker LH,
Bonadonna G. Phase II evaluation of adriamycin in human neoplasia.
Cancer. 1973;32(1):1-8.
54. Antman KH. Adjuvant therapy of
sarcomas of soft tissue.
Semin Oncol. 1997;24(5):556-60.
55. Adjuvant chemotherapy for
localised resectable softtissue sarcoma of adults: meta-analysis of
individual
data. Sarcoma Meta-analysis Collaboration. Lancet.
1997;350(9092):1647-54.
56. Pervaiz N, Colterjohn N,
Farrokhyar F, Tozer R, Figueredo A, Ghert M. A systematic meta-analysis
of randomized controlled trials of adjuvant chemotherapy
for localized resectable soft-tissue sarcoma. Cancer.
2008;113(3):573-81.
57. Mouridsen HT, Bastholt L, Somers
R, Santoro A, Bramwell V, Mulder JH et al. Adriamycin versus epirubicin
in advanced soft tissue sarcomas. A randomized
phase II/phase III study of the EORTC Soft Tissue
and Bone Sarcoma Group. Eur J Cancer Clin Oncol.
1987;23(10):1477-83.
58. Judson I, Radford JA, Harris M,
Blay JY, van Hoesel Q,
le Cesne A et al. Randomised phase II trial of pegylated
liposomal doxorubicin (DOXIL/CAELYX) versus doxorubicin in the
treatment of advanced or metastatic soft
tissue sarcoma: a study by the EORTC Soft Tissue and
Bone Sarcoma Group. Eur J Cancer. 2001;37(7):870-7.
59. Patel SR, Vadhan-Raj S, Burgess
MA, Plager C, Papadopolous N, Jenkins J et al. Results of two
consecutive
trials of dose-intensive chemotherapy with doxorubicin
and ifosfamide in patients with sarcomas. Am J Clin Oncol.
1998;21(3):317-21.
60. Gottlieb JA, Benjamin RS, Baker
LH, O'Bryan RM,
Sinkovics JG, Hoogstraten B et al. Role of DTIC (NSC45388) in the
chemotherapy
of sarcomas. Cancer Treat
Rep. 1976;60(2):199-203.
61. Garcia-Del-Muro X, Lopez-Pousa A,
Maurel J, Martin J,
Martinez-Trufero J, Casado A et al. Randomized phase
II study comparing gemcitabine plus dacarbazine versus
dacarbazine alone in patients with previously treated
soft tissue sarcoma: a Spanish Group for Research on
Sarcomas study. J Clin Oncol. 2011;29(18):2528-33.
62. Maki RG, Wathen JK, Patel SR,
Priebat DA, Okuno SH,
Samuels B et al. Randomized phase II study of gemcitabine and docetaxel
compared with gemcitabine alone in
patients with metastatic soft tissue sarcomas: results of
sarcoma alliance for research through collaboration study 002
[corrected]. J Clin Oncol. 2007;25(19):2755-63.
63. Demetri GD, von Mehren M, Jones
RL, Hensley ML,
Schuetze SM, Staddon A et al. Efficacy and Safety of
Trabectedin or Dacarbazine for Metastatic Liposarcoma
or Leiomyosarcoma After Failure of Conventional Chemotherapy: Results
of a Phase III Randomized Multicenter Clinical Trial. J Clin Oncol.
2016;34(8):786-93.
64. Schoffski P, Chawla S, Maki RG,
Italiano A, Gelderblom
H, Choy E et al. Eribulin versus dacarbazine in previously treated
patients with advanced liposarcoma or
leiomyosarcoma: a randomised, open-label, multicentre,
phase 3 trial. Lancet. 2016;387(10028):1629-37.
65. van der Graaf WT, Blay JY, Chawla
SP, Kim DW, BuiNguyen B, Casali PG et al. Pazopanib for metastatic
soft-tissue sarcoma (PALETTE): a randomised,
double-blind, placebo-controlled phase 3 trial. Lancet.
2012;379(9829):1879-86.
Recibido:
Octubre 26, 2020
Aprobado: : Octubre 30, 2020
Correspondencia:
Anna Estival
aestival@iconcologia.net