Resumen
La comprensión de que la progresión del cáncer requería la interacción
de múltiples genes proporcionó
una de las razones fundamentales, para embarcarse en 1986, en el
proyecto genoma humano. Solo con
una secuencia del genoma de referencia podría entenderse el espectro
completo de cambios somáticos
que conducen al cáncer. Desde su finalización en 2003, la secuencia del
genoma humano de referencia
ha cumplido su promesa como herramienta fundamental para esclarecer la
patogénesis de diversas
neoplasias. Los recientes avances biotecnológicos han llevado a la
identificación de características
biológicas complejas y únicas asociadas con la carcinogénesis. La
perfilación del ADN tumoral libre
circulante y de las células neoplásicas, así como de factores
relacionados con inmunidad, análisis de
proteinas y del ARN, permiten optimizar el diagnóstico y tratamiento
del cáncer. En consecuencia, la
búsqueda de respuestas con base en experimentos clínicos ha
evolucionado, pasando de los estudios
centrados en un tipo específico de tumor a una o muchas características
genómicas, independientes de
la histología, y con base en diseños innovadores y adaptativos. A
continuación, revisamos los hitos y
conceptos históricos clave en la genómica del cáncer y algunos de los
descubrimientos novedosos en
esta área.
Palabras clave: Cáncer; genómica; medicina de precisión;
biomarcador; ADN; mutación
CANCER GENOMICS EVOLUTION
Abstract
The realization that cancer progression required the interaction of
multiple genes provided
one of key rationales, in 1986, for embarking on the human genome
project. Only with a reference genome sequence could the full spectrum
of somatic changes leading to cancer be
understood. Since its completion in 2003, the human reference genome
sequence has fulfilled
its promise as a foundational tool to illuminate the pathogenesis of
diverse neoplasms. In recent years, biotechnological breakthroughs have
led to identification of complex and unique
biologic features associated with carcinogenesis. Tumor and cell-free
DNA profiling, immune
markers, and proteic and RNA analyses are used to identify these
characteristics for optimization of anticancer therapy in individual
patients. Consequently, clinical trials have evolved,
shifting from tumor type-centered to gene-directed, histology-agnostic,
with innovative adaptive design tailored to biomarker profiling with
the goal to improve treatment outcomes. Herein,
we review the key historical milestones and concepts in cancer
genomics, and some of the
novel discoveries in this area.
Keywords: Cancer;
genomics; precision medicine; biomarker; DNA; mutation

¹ MD MSc PhD. Fundación para la Investigación Clínica y
Molecular Aplicada del Cáncer – FICMAC, Bogotá, Colombia.
² Grupo Oncología Clínica y Traslacional, Clínica del Country,
Bogotá, Colombia.
³ Grupo de Investigación en
Oncología Molecular y Sistemas
Biológicos (FoxG), Universidad El Bosque, Bogotá, Colombia.
⁴ Sección Oncología Torácica y
Laboratorio de Medicina
Personalizada del Cáncer, Instituto Nacional de Cancerología – INCaN,
Ciudad de México, México.
⁵ Departamento Oncología Clínica, Marlene and Stewart Greenebaum
Comprehensive Cancer Center, Facultad de Medicina Universidad de
Maryland, Baltimore, Maryland, Estados Unidos.
⁶ MD. Fundación para la Investigación Clínica y
Molecular Aplicada del Cáncer – FICMAC, Bogotá, Colombia.
Introducción
En 1971, la clonación del ADN recombinante, la técnica más novedosa
para la época, estaba todavía en
su infancia y aún no era capaz de lidiar con la tarea
de desentrañar toda la complejidad de la célula humana. Sin embargo, no
fue sino hasta 1985 cuando
Dulbecco presentó la prueba de que las mutaciones
en los genes podrían causar cáncer, un hecho que hoy
damos por sentado, brindando el valor de la secuencia
genómica como base para comprender la enfermedad
(1). La discusión contemporánea sobre la generación
de la secuencia humana completa representó un salto asombroso ya que,
en ese momento, secuenciar un
solo gen de aproximadamente 1 kilobase era digno de
una tesis, y el genoma humano era 3 millones de veces
más grande. La estructura repetida del genoma estaba
bien caracterizada y algunos también la consideraban
un obstáculo insuperable para tal esfuerzo y una base
para la resistencia temprana al proyecto (2). A pesar
de estos obstáculos, un esfuerzo internacional condujo a la
finalización del genoma de referencia humano
en 2003 (International Human Genome Sequencing
Consortium 2004), y con esta reseña completa, fue posible probar el
valor de la genómica para descifrar los
cambios que conducen al cáncer. Los datos que han
surgido desde 2003 respaldan de manera abrumadora
el valor de esta visión y han cambiado la forma en que
se investiga y se comprenden las neoplasias.
Durante el período de secuenciación del genoma humano (1990-2003) (
http://www.ornl.gov/sci/techresources/Human_Genome/project/about.shtml),
la investigación continuó acumulando conocimiento sobre la
clonación, permitiendo mejorar la capacidad de secuenciación lo que
llevó a identificar la mayoría de los oncogenes y genes supresores de
tumor. Un inventario de
las alteraciones asociadas al cáncer determinó la presencia de 291
genes conductores, lo que corresponde
al ~1% de la secuencia codificante (3). Los primeros
estudios en profundidad observaron que el 90% de
estos genes presentaban mutaciones somáticas, cerca del 20% tenía
alteraciones germinales, y el 10%
contenía ambas categorías. Las formas más comunes
de variación en el repertorio mutaciones contenido
hasta 2004, fueron las traslocaciones que conducían a
la generación de proteínas de fusión con carácter oncogénico. Hasta ese
año, nadie había estudiado más
de un puñado de genes tumorales, lo que representó
el cambio más significativo en el conocimiento para
la oncología actual.
Ese fue el estado de la investigación en el umbral de la
“era genómica” del cáncer, un tiempo anunciado por
la disponibilidad del genoma de referencia y la espectacular explosión
de datos, alimentado por la introducción de sistemas operativos para el
control masivo de
información y el desarrollo de instrumentos de secuenciación paralela
más económicos. Este año es el vigésimo aniversario de la publicación
de ese notable hito
en la ciencia: la finalización del genoma humano de
referencia. En esta coyuntura, recapitulamos algunos
de los hallazgos y desafíos clave que han surgido del
análisis de la secuencia del genoma del cáncer.
Nomenclatura de las alteraciones
genómicas
La genómica, en lo que respecta al cáncer, es el estudio de cómo las
alteraciones en el código genético dan
lugar a células anormales. Aunque ciertamente no está
tan rígidamente instalado como antes, es importante
considerar el dogma central de la biología molecular
que describe el flujo jerárquico de información genética desde el ADN
hasta la proteína. La información genética codificada en el ADN se
transcribe a ARN mensajero (ARNm) por la ARN polimerasa, y el ARNm
se traduce posteriormente a la proteína a través de los
ribosomas, lo que da como resultado una expresión
fenotípica del gen subyacente. Este proceso de transferencia de
información del ADN a la proteína funcional se conoce como expresión
génica. Casi el 90%
del ADN genómico humano se transcribe en ARN;
sin embargo, solo del 1% al 2% de las transcripciones se expresan como
proteínas. La mayoría de estas
transcripciones son ARN no codificantes (ncRNA),
que apenas estamos comenzando a reconocer por su
importancia como reguladores de las funciones celulares. Los errores en
el procesamiento y transferencia de
información genética conducen al desarrollo de mutaciones que dan lugar
a una variabilidad significativa en
el genoma pudiendo llegar a la génesis del cáncer (4).
El ADN es la unidad básica de información de todos
los organismos. Está compuesto de nucleótidos purina
(adenina, guanina) y pirimidina (citosina, timina). Estos se organizan
en secuencias para formar genes, que
son las unidades estructurales y funcionales de información hereditaria
dispuestas por combinaciones variadas de los cuatro nucleótidos.
El ADN se envuelve
alrededor de las histonas y se agrupa como cromatina
dentro del núcleo celular. Este material genético está
organizado en cromosomas que se transportan por duplicado (diploide)
con la excepción de los cromosomas
sexuales, en la combinación XY. Estas dos versiones
separadas del mismo gen en las células diploides se
conocen como alelos, que se encuentran en forma homocigótica (dos
alelos idénticos) o heterocigótica (dos
alelos diferentes). Los cambios en esta secuencia de
ADN afectan potencialmente la función y el desarrollo de un organismo
(5).
Las mutaciones son omnipresentes en las células
neoplásicas (6). Estos cambios en la secuencia de
ADN pueden conducir a una expresión o función
proteica alterada. Hay múltiples tipos que pueden estar relacionadas
con el desarrollo del cáncer, y estas
ocurren a través de varios mecanismos, que incluyen agresiones
extrínsecas y defectos intrínsecos. Las agresiones extrínsecas o
mutágenos, son agentes naturales
o artificiales que producen alteraciones en la secuencia
del ADN. Estos mutágenos pueden ser biológicos (por
ejemplo, virus de Epstein-Barr o el virus del papiloma
humano), químicos (por ejemplo, bromuro de etidio,
etc.) o radiológicos (por ejemplo, radiación ultravioleta [UV],
radiación ionizante). Los defectos intrínsecos incluyen fallas en la
replicación o reparación del
ADN. Dependiendo del tipo de células en las que ocurren estas
mutaciones, se clasifican en línea germinal
(heredables y en células reproductoras) o somáticas
(no heredables y no reproductivas). La acumulación
de mutaciones es un factor clave y responsable del desarrollo,
crecimiento, metástasis y resistencia al tratamiento del cáncer (4).
Las mutaciones a menudo se clasifican según el grado
de alteración del ADN. Por ejemplo, las mutaciones
puntuales o pequeños cambios suelen afectar a un solo
gen, mientras que las anomalías cromosómicas alteran
grandes segmentos del ADN. Las mutaciones puntuales son el resultado de
sustituciones, inserciones o
deleciones de un solo par de bases. Las sustituciones
de bases pueden provocar transiciones o transversiones. Se produce una
transición cuando una pirimidina
se cambia por una pirimidina diferente o una purina
se cambia por una purina alternativa después de una
sustitución de base, y una transversión, ocurre cuando
una pirimidina se convierte en una purina o viceversa. Si estas
alteraciones resultan en la sustitución de
un aminoácido diferente al esperado en una secuencia
codificante para una proteína, se conoce como una
mutación de cambio de sentido o missense como es
conocida en inglés. Las inserciones o deleciones (indels) pueden dar
como resultado desplazamiento en el
marco en las que la lectura se desplaza de modo que
se altera la secuencia de aminoácidos que se está codificando (
frameshift). Si el marco de
codificación de
aminoácidos es alterado por cualquier mutación de
una manera que conduce a la introducción prematura
de un codón de terminación (parada), resulta en una
proteína truncada. Estas mutaciones, al igual que las
puntuales que ocasionan el mismo resultado se conocen como mutaciones
sin sentido (
non-sense).
Las grandes alteraciones del ADN, como las anomalías cromosómicas o
rearreglos, modifican la estructura de los cromosomas a través de la
pérdida,
la ganancia o el reordenamiento de segmentos. Estas
anomalías ocurren cuando los cromosomas sufren
procesos de ruptura y reunión, lo que resulta en el intercambio o la
pérdida de material genético dentro del
mismo o entre diferentes cromosomas (
Figura
1). Si
bien algunas anomalías cromosómicas son hereditarias, muchas ocurren de
manera espontánea o como
consecuencia de factores ambientales como la exposición a medicamentos,
rayos X, rayos UV, u otros factores carcinogénicos. Hay cuatro
rearreglos estructurales
cromosómicos comunes que incluyen las inversiones,
deleciones, duplicaciones y translocaciones (4).
Las inversiones ocurren cuando el segmento de un cromosoma se desprende
y luego se vuelve a unir en la
orientación inversa al mismo cromosoma (7). Como
sugiere el nombre, una deleción es la pérdida de un
segmento cromosómico (como en el síndrome de Cridu-chat) de manera que
el material genético perdido
no se puede recuperar. La duplicación de un segmento
cromosómico puede ocurrir cuando una parte del cromosoma se copia y se
inserta junto al segmento original en tándem o en orientación inversa
(7). Las translocaciones implican la transferencia de un segmento
de un cromosoma a una parte diferente del mismo cromosoma o a un
cromosoma completamente diferente.
Genéticamente, sobre todo si los puntos donde se da
la ruptura o la reunión comprometen regiones críticas, puede dar como
resultado el cambio de secuencia,
posición y orden de algunos genes, lo que a menudo
en cáncer, resulta en varias anomalías funcionales que
implican una consecuencia biológica. La leucemia
mielógena crónica (LMC), una de las neoplasias relacionadas con una
translocación mejor caracterizadas,
se inicia cuando la porción del cromosoma 9 que alberga el código
genético de la tirosina-quinasa, el gen
de la
leucemia murina de Abelson
(ABL), se une al gen
de la proteína de la
región de
clúster de punto de ruptura
(BCR) ubicada en el cromosoma 22, formando el gen
de fusión BCR-ABL, conocido como cromosoma Filadelfia. El producto
proteico de este rearreglo, exhibe
una actividad tirosina-quinasa patológicamente incrementada, fomentando
la proliferación celular. Conociendo este mecanismo esta translocación
se convirtió
en el objetivo de algunas de las primeras terapias contra el cáncer
dirigidas molecularmente (8).
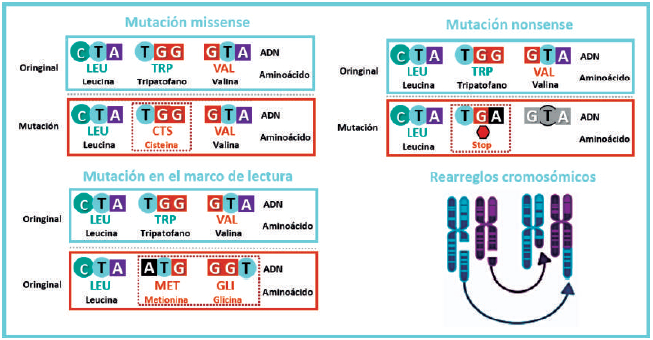
Figura 1. Clasificación de las mutaciones en cáncer.
Además de las variaciones estructurales, también se
producen aberraciones numéricas de los cromosomas
que se denominan aneuploidía (una pérdida o una ganancia de uno o más
cromosomas) y polisomías (uno
o más genomas completos en una célula que pueden
ser idénticos o distintos entre sí) (7). Estos se han asociado con el
desarrollo de algunas neoplasias como el
neuroblastoma, los astrocitomas pediátricos y los condrosarcomas
(9-11).
Modificaciones epigéneticas
El término epigenética denota cambios en el fenotipo
de una célula sin afectar el genotipo, o la secuencia
de la misma (12). Más recientemente, la epigenética se
ha definido como todos los cambios hereditarios en la
expresión génica y en la estructura de la cromatina que
no están codificados en la secuencia propia del ADN
(5). En todos los organismos multicelulares eucariotas, los procesos de
diferenciación están controlados
a través de estos mecanismos, con la excepción de la
maduración de las células T y B (5). La regulación
epigenética de los fenotipos incluye la metilación del
ADN, las modificaciones de histonas y la expresión
de micro-ARN (miARN). Los cambios en cualquiera de estos reguladores
epigenéticos pueden provocar
una alteración de la expresión génica y el desarrollo de
cáncer y otras enfermedades crónicas (5).
La metilación del ADN es la adición de un grupo metilo a las citosinas
ubicadas en secuencias de ADN
conocidas como islas CpG (repeticiones de citosina
seguida de guanina). Es una de las primeras modificaciones epigenéticas
descritas, y aproximadamente
del 70% al 80% de las citosinas en estas estructuras
están metiladas en células somáticas humanas (13). La
adición de grupos metilo está mediada por un complejo de enzimas
catalíticas conocidas como ADN metiltransferasas (DNMT). Para
contrarrestar la acción
de las DNMT, las demetilasas eliminan estos grupos
metilo, resultando en un balance homeostático de activación e
inactivación de transcripción. Es entonces
que los procesos de metilación del ADN juegan un
papel crucial en el mantenimiento de la estabilidad
del genoma, la inactivación del cromosoma X femenino, así como en la
regulación de la expresión génica
y los procesos de desarrollo (14,15). Las alteraciones
en estos patrones fueron las primeras alteraciones epigenéticas
identificadas en el cáncer. En las neoplasias
se han identificado fenómenos de hipometilación, una
disminución general de los patrones de metilación del
ADN, así como la hipermetilación, un aumento global de la metilación de
las islas CpG (15). Un ejemplo
del segundo se ha identificado en los promotores del
gen CDKN2A, el cual lleva a la pérdida de la expresión
de los supresores tumorales p16 y p14arf y por ende,
promueve el proceso oncogénico por pérdida de tan
importantes reguladores del ciclo celular (16). Alternativamente, la
hipometilación se ha identificado especialmente a nivel de los
promotores de protooncogenes
y factores de crecimiento, fomentando la proliferación
e inmortalización de células malignas (15).
Otro cambio epigenético importante que puede conducir al cáncer implica
la modificación postraduccional de las histonas. Vale la pena
mencionar, que, al
contener la cadena de ADN empaquetada, los cambios que involucren
especialmente residuos que interactúan por diversas fuerzas
electroquímicas sobre la
misma, van a repercutir directamente en esta relación,
fomentando o inhibiendo la condensación de la hebra.
Los principales cambios sobre estas moléculas son la
metilación, acetilación, fosforilación, ubiquitilación y
sumoilación. La metilación de regula varios procesos
celulares como el procesamiento del ARN, la biosíntesis de ribosomas,
la reparación del ADN, la transducción de señales y la regulación
transcripcional. Se
sabe adicionalmente que la metilación sobre residuos
lisina/arginina es directamente responsable de la activación o
represión transcripcional (5). La acetilación
y deacetilación, que están mediadas por enzimas conocidas como histonas
acetiltransferasas (HAT) e
histonas deacetilasas (HDAC), respectivamente, son
fundamentales para regular la dinámica de la cromatina, la
transcripción, el silenciamiento de genes, la
progresión del ciclo celular, la apoptosis, la diferenciación, la
replicación del ADN y la reparación del mismo.
Un de-sequilibrio en la ponderación de la acetilación y
desacetilación de histonas está directamente relacionado
con la tumorigénesis y la progresión de la enfermedad. Se
han observado alteraciones de los genes que codifican
las enzimas HAT y HDAC en leucemias y cánceres de
colon, útero y pulmón, y se han desarrollado de este
proceso para atacar estas aberraciones en las células
cancerosas (12).
La fosforilación de histonas es menos común en comparación con la
acetilación y metilación. Sin embargo,
este proceso sobre un residuo de serina inicia la respuesta al daño del
ADN y sobre una treonina controla la estructura de la cromatina
mediante la señalización de las
proteínas modificadoras de marcas epigenéticas (12).
Los ncARN, como el miARN y el ARN de interferencia corta (siARN), se
han identificado como reguladores clave adicionales de la expresión
génica. Al
interferir con procesos como la traducción de ARNm,
la metilación del ADN y las modificaciones de histonas, los miARN
regulan procesos celulares como
el desarrollo, la proliferación, la diferenciación y la
muerte celular. Estos miARN están constituidos por
20 a 22 nucleótidos de longitud y están ubicados en
sitios frágiles que son susceptibles a amplificaciones, o
ganancia en el número de copias. Diferentes estudios
han mostrado evidentes diferencias en los perfiles de expresión de
miARN en tejidos normales frente a los
tumorales. Adicionalmente, estos afectan a los genes
supresores tumorales, los oncogenes y los genes de reparación del ADN
(17). Datos recientes han demostrado una utilidad emergente de los
miARN como herramientas en el diagnóstico, pronóstico y tratamiento del
cáncer (18), pero aún queda mucho por dilucidar con
respecto a la diversidad de funciones que desempeñan
estos reguladores epigenéticos.
Mecanismos de reparación del ADN
Las células son desafiadas constantemente por agresiones al genoma que
podrían resultar en mutaciones.
Existen varias vías de reparación y respuesta al daño
del ADN para reconocer estos efectos relativamente
comunes, y mantener la estabilidad e integridad genómica. Cuando estos
procesos fallan es que puede
originarse las diferentes alteraciones mencionadas
previamente, iniciando el proceso oncogénico. Dentro
de los mecanismos involucrados en este proceso están
la reparación por escisión de bases, la reparación por
escisión de nucleótidos, la reparación por desajustes
(MMR), y la reparación por escisión bicatenaria (19).
El primero hace referencia a la corrección de una o
dos bases las cuales se encuentran alteradas, generalmente por cambios
químicos tales como oxidaciones
que a su vez no modifican la estructura helicoidal de
la cadena. Este mecanismo retira la base afectada al
igual que un pequeño fragmento adyacente el cual es
reparado por medio de una polimerasa. En caso de
que haya distorsión de la estructura de la hélice, ya sea
por radiación ultraviolenta, mutágenos ambientales o
exposición a agentes quimioterapéuticos, a menudo se
elimina mediante reparación por la vía NER o de escisión de
nucleótidos. Los defectos en este mecanismo
de reparación se asocian con envejecimiento prematuro y el cáncer. NER
consiste en el reconocimiento del
daño, el desenrollamiento del ADN alrededor de la
lesión, la escisión y la modificación de la hebra alterada para generar
la síntesis del nuevo ADN incluido
con una ligadura final simultánea (20). La reparación
por la vía NER defectuosa relacionada con el xeroderma pigmentoso (XP)
da como resultado la presentación de múltiples cánceres de piel
inducidos por rayos
UV (20). Esta es una rara enfermedad hereditaria con
carácter autosómico recesivo en la que el afectado
muestra una marcada tendencia a desarrollar lesiones
neoplásicas cutáneas dependientes de la exposición al
sol; los heterocigotos son frecuentemente asintomáticos y no
desarrollan la enfermedad. La lesión más
significativa que produce la luz ultravioleta sobre el
ADN consiste en la formación de dímeros de timina, es decir, dos
timinas adyacentes (en una misma
cadena de ADN) las cuales se unen covalentemente,
causando alteraciones en el proceso de replicación
del ADN. En humanos, varios complejos enzimáticos
(fotoliasas y su cofactor FADH2) se hallan implicados en la reparación
de este fenómeno por medio de
un complejo enzimático que absorbe la luz y utiliza
esta energía para romper el enlace que une el dímero,
separando las dos timinas. Es entonces que esta enfermedad es
consecuencia de la mutación de cualquiera
de los siete genes implicados en este mecanismo de
reparación (21).
El sistema MMR (del inglés mismatch repair) mantiene la estabilidad
genómica corrigiendo las bases mal
emparejadas formadas debido a la mutación, sustitución, deleción o
inserción de nucleótidos. El MMR
ocurre durante la fase S del ciclo celular en donde se
da una corrección por un proceso de escisión de la
base anómala e inserción de la correcta. Algunas de las
proteínas clave necesarias para el correcto funcionamiento del sistema
MMR son MSH2, MLH1 y ADN
polimerasas δ y ε. Las mutaciones en MSH2 y MLH1
están relacionadas con un fenotipo hipermutado y con
la inestabilidad del genoma, evento relativamente frecuente en el
cáncer de colon (22). Durante una fase de
replicación eficaz, las ADN polimerasas δ y ε son necesarias para la
actividad de corrección y, si se produce
una mutación en estas enzimas, los desajustes de las bases aumentan,
dando lugar a mutaciones complejas
(23,24). La pérdida de la función de la vía de reparación MMR está
relacionada con cánceres esporádicos
y hereditarios (25).
Las roturas de doble cadena del ADN son lesiones
considerablemente lesivas y están relacionadas con
la pérdida de grandes regiones cromosómicas, lo que
conduce a un desgaste de la integridad genómica. La
falta de reparación de las roturas de doble hebra a menudo conduce a la
pérdida de información genética,
inestabilidad y fragmentación del genoma, reordenamiento cromosómico y
muerte celular (25). Una célula
de mamífero emplea dos vías diferentes para restaurar
las rupturas bicatenarias: recombinación homóloga
y unión de extremos no homólogos. La recombinación homóloga requiere
homología de la secuencia de
ADN para reparar la lesión de ADN y está activa en
las fases media S y G2 del ciclo celular. Los cánceres
hereditarios de seno y ovario están asociados con mutaciones en los
genes
BRCA1 o BRCA2. Ambas
proteínas BRCA son necesarias para la reparación de roturas
bicatenarias del ADN competente mediante recombinación homóloga. La
unión o reparación de extremos
no homólogos no requiere paralelismo de secuencia
para reparar roturas de doble cadena y es activa durante todo el ciclo
celular, pero es dominante en las fases
G0/G1 y G2 (24,25).
Además de los mecanismos de reparación mencionados existe una serie de
componentes que revierten
directamente algunas lesiones del ADN en un proceso de un solo paso
denominado reversión directa. Por
ejemplo, el daño inducido por los rayos UV se repara mediante enzimas
conocidas como fotoliasas. Las
lesiones oxidativas son reparadas directamente por la
enzima metilguanina-metiltransferasa (MGMT) (24).
Los datos clínicos emergentes proporcionan evidencia
de que la sobreexpresión de los factores de reparación
del ADN puede tener un significado pronóstico y predictivo en diversas
neoplasias, y la inhibición de la
reparación del ADN se ha convertido en un objetivo
prometedor para el cáncer de seno, próstata y ovario.
Métodos básicos para genotipificación en
cáncer
Tradicionalmente, los tipos de cáncer se han definido y asociado con la
edad de aparición, el origen
anatómico, la histología, y las características inmunohistoquímicas.
Actualmente, se emplean varias técnicas moleculares para detectar
aberraciones genéticas,
como la PCR, la secuenciación Sanger, la secuenciación de próxima
generación (NGS), la hibridación por
fluorescencia in situ (FISH), y otros (26). La elección
del método de detección utilizado depende de una
variedad de factores, como el tipo de muestra (tejido,
sangre, fluidos), volumen de la muestra, genes a analizar y tipo de
mutaciones en el gen de interés (27). La
PCR o reacción de cadena de polimerasa, por sus siglas en inglés, es
una técnica molecular de uso común
para amplificar directamente segmentos específicos de
ADN mediante ciclos repetidos de desnaturalización,
hibridación y alargamiento. El producto amplificado
se puede utilizar posteriormente para diversas aplicaciones. La PCR
alelo-específica es un método basado
en tiempo real que detecta mutaciones conocidas con
alta sensibilidad. Requiere dos sondas fluorescentes,
una para el alelo de tipo salvaje y otra para el alelo
mutante. Puede detectar ADN mutante incluso si el
contenido de tumor en una muestra es solo del 1 a 5%.
Por otro lado, y similar a su contraparte previamente mencionada, la
PCR de microfluido digital (digital
droplet PCR, ddPCR) se usa para detectar mutaciones
conocidas, incluida las variaciones de un solo nucleótido, pequeñas
inserciones, deleciones, y reordenamientos con alta sensibilidad y
especificidad. La ddPCR
permite un análisis más preciso y cuantitativo que la
PCR convencional o en tiempo real. Su función en
la detección de mutaciones en el ctADN (circulating
tumor DNA) ha sido esencial en el desarrollo de la
biopsia líquida, como estrategia diagnóstica y para el seguimiento. A
diferencia de la PCR, la secuenciación
Sanger se utilizó para detectar nuevas alteraciones.
Esta técnica utiliza un producto de PCR para detectar mutaciones nuevas
o establecidas, incluida la variación de un solo nucleótido, pequeñas
duplicaciones,
inserciones y deleciones, pero no detecta cambios en el
número de copias. Requiere que el ADN mutante esté
presente en al menos el 20 a 25% de las células de la
muestra. La lógica detrás de esta radica en replicar los
productos de la PCR por medio de una polimerasa.
Con el fin de determinar que nucleótido es incluido se
emplean didesoxinucleótidos (ddNTP) marcados con
un tinte fluorescente único, los cuales al carecer de un
grupo hidroxilo en la posición 3´del anillo de ribosa,
inhiben el proceso de replicación. De esta manera, se
generan fragmentos de diferentes tamaños cuyo extremo contiene
precisamente ese marcaje fluorescente. Al
separar estos fragmentos de ADN por tamaño y leer el
tinte fluorescente cuando cada uno pasa por un detector, los
nucleótidos se pueden leer de forma secuencial
con una alta precisión, estableciendo la secuencia (27).
Debido a la complejidad del proceso, los costos y la velocidad de
reacción se desarrollaron nuevas técnicas,
conocidas como secuenciación de próxima generación.
Las plataformas de NGS permiten una secuenciación
profunda y paralela masiva de cadenas de oligonucleótidos durante una
sola serie de secuenciación, necesitando menos material para el
análisis, lo que es útil
para los entornos tumorales donde la muestra de tejido
es limitada. Se basa en la fragmentación del ADN; sin
embargo, en lugar de requerir una configuración separada para cada
reacción como en la secuenciación
Sanger, la NGS utiliza bibliotecas de ADN molde fijadas en una
superficie que permite la amplificación
por PCR paralela a gran escala con una cantidad relativamente pequeña
de reactivo. Dependiendo de la
plataforma NGS, se utilizan varios mecanismos bioquímicos diferentes
para determinar la secuencia del
fragmento. La NGS puede detectar simultáneamente
sustituciones de bases únicas, duplicaciones, inserciones, deleciones,
y variaciones en el número de copias
de múltiples genes en un solo ensayo. Al considerar
los resultados de los estudios actuales, es importante
comprender que se puede realizar estudios del genoma
completo, del exoma (la totalidad del material codificante) o genes
específicos de interés en muestra tumorales. La interpretación de estos
hallazgos es un poco
más compleja que las otras técnicas debido a que por
la química de reacción pueden introducirse artefactos
que pueden ser anómalamente interpretados como
mutaciones con impacto sobre la biología tumoral. Sobre este aspecto al
igual que analizar la gran cantidad
de datos involucrados en obtener secuencias de forma
masiva, se requirieron grandes avances en la bioinfomática y biología
computacional.
Para la detección de rearrelos o cambios en el número de copias, el
FISH es un buen método diagnóstico.
Este utiliza sondas marcadas con fluorescencia unidas
a secuencias de interés los cuales se van a hibridar con
su región correspondiente en el genoma a indagar. En
el caso de obtener aumento de las señales de fluorescencia sobre lo
esperado, se podría estimar una ganancia de número de copias. Por otro
lado, las pérdidas
de información se traducirían en ausencia de la señal
de interés. Adicionalmente, mediante el diseño de sondas específicas
como sondas de fusión o de ruptura,
se pueden identificar estos tales como el cromosoma
Filadelfia (27).
Las micromatrices de ADN (DNA microarrays) o microarreglos se utilizan
para identificar múltiples alteraciones conocidas, usando un chip
sólido que contiene
miles de cadenas simples de ADN para hibridar con
la muestra de ADN del tumor. Es importante destacar que el ADN
monocatenario es sintético y puede
prepararse para cualquier secuencia de ADN deseada,
por tanto, se puede crear una matriz con diferentes
objetivos de interés. La muestra de ADN se etiqueta
con un tinte fluorescente y se hibrida con las hebras de
ADN del chip. Se utiliza detección fluorescente para la detección y
cuantificación, más un sistema operativo
especializado para determinar la expresión, cantidad
y secuencia de la muestra de ADN proveniente del tumor (27).
Alteraciones genómicas más
frecuentes
La frecuencia media de mutaciones puntuales varía
en más de tres órdenes de magnitud en los cánceres
más comunes; dentro de un tipo de tumor particular,
la variación en la frecuencia mutacional es de un orden
de magnitud (
Figura 2). La
variación en la frecuencia de las mutaciones es una función del número
de
divisiones de las células somáticas antes de la iniciación del tumor.
En el extremo inferior de la escala se
encuentran las neoplasias pediátricas, seguidas de las
leucemias agudas en adultos y de los tumores sólidos.
Los tumores que superan las 10 mutaciones por Megapar de bases (Mbp) a
menudo tienen deficiencias
en la reparación de errores propios del apareamiento,
ya sea por una mutación o silenciamiento epigenético
(por ejemplo, MLH1). Los tumores con más de 100
mutaciones por Mbp suelen tener mutaciones en la
exonucleasa POLE, una de las dos enzimas de replicación del ADN (The
Cancer Genome Atlas Research
Network). Estos patrones pueden tener implicaciones
importantes para la clínica, como ocurre para los pacientes con
carcinoma colorrectal que tienen disfunción replicativa por daños en la
reparación, evento
que condiciona un mejor pronóstico en especial tras
la exposición a la inmunoterapia. En el otro extremo
de la escala, se encuentran la mayoría de los tumores
pediátricos que presentan pocas mutaciones, hallazgo
que limita las posibilidades terapéuticas.
El censo inicial de los genes relacionados con el desarrollo del cáncer
dio paso a COSMIC, el Catálogo de
mutaciones somáticas en el cáncer (28). En los tumores sólidos más
comunes un promedio de 33 a 65 genes presenta mutaciones somáticas que
modifican sus
productos proteicos. Aproximadamente el 95% de estas
mutaciones son sustituciones de una sola base (como
C> G), mientras que el resto son deleciones o inserciones de una o
pocas bases (como CTT> CT). De las
sustituciones de bases, el 91 % dan como resultado cambios sin sentido,
el 7,6% dan como resultado cambios
sin sentido, y el 1,7% generan alteraciones de los sitios
de empalme o regiones no traducidas inmediatamente
adyacentes a los codones de inicio y finalización. Información
actualizada ha permitido elucidar que la mejor
manera de identificar genes mutados que actúan como
conductores es valorando el patrón mutacional en lugar
de su frecuencia de alteración. Los patrones de mutaciones entre los
oncogenes y genes supresores de tumores
bien estudiados son característicos y no aleatorios; los
oncogenes mutan de manera recurrente en las mismas
posiciones de aminoácidos, mientras que los genes supresores de tumores
mutan a través de alteraciones que
truncan proteínas en toda su longitud. Sobre la base
de estos patrones de mutación en lugar de frecuencias,
ha sido posible determinar cuáles de los 18.306 genes
mutados que contienen un total de 404.863 alteraciones registradas en
COSMIC actúan como conductores.
Para ser clasificado como un oncogén es necesario que
más del 20% de las mutaciones registradas en el gen estén en posiciones
recurrentes y no tengan sentido. Por
otra parte, para ser clasificado como un gen supresor
de tumores, análogamente se requiere que más del 20%
de las mutaciones registradas en el gen sean inactivadoras. Esta regla
20/20 es indulgente en el sentido de que
todos los genes del cáncer bien documentados superan
con creces estos criterios (4,29).
El inventario más reciente de mutaciones en cáncer incluidas en COSMIC (
http://www.sanger.ac.uk/genetics/CGP/Census/)
incluye más de 900.000 eventos
somáticos.
Sin embargo, hasta la fecha solo se han descubierto
125 genes con mutaciones conductoras, 71 son genes
supresores de tumor y 54 son oncogenes (4). Cada tipo de neoplasia
tiene una colección característica de genes alterados, como lo
ejemplifica el cáncer colorrectal (Figura 3).
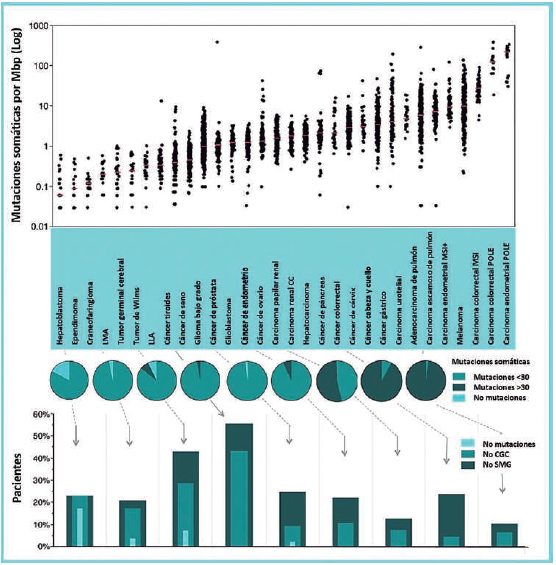
Figura 2. Frecuencias de mutaciones somáticas en pacientes con
cáncer. Todos los datos representan tumores
primarios. Solo se contaron las mutaciones deletéreas (sin sentido, por
desplazamiento del marco y sitio de
empalme). Arriba se observan las frecuencias generales de las
mutaciones somáticas donde cada punto negro
representa un tumor. El grupo sombreado en azul claro denomina los
tumores pediátricos y el sombreado en azul
profundo indica las neoplasias de los adultos. Las líneas horizontales
rojas dentro de cada grupo de puntos indican
la mediana para el valor de la frecuencia de mutaciones de cada
neoplasia. (LLA) Leucemia linfoide aguda; (LMA)
leucemia mieloide aguda; (CRC) carcinoma colorrectal; (MSI)
inestabilidad microsatélital; (POLE) pacientes con
mutaciones somáticas en el dominio de nucleasas del gen POLE. En la
porción inferior de la figura se observa
la clasificación de frecuencia de los tumores por mutación. Los
gráficos circulares dividen a los pacientes en tres
grupos según la frecuencia de la mutaciones deletéreas: 0 mutaciones
somáticas detectables, menos de 30, y
mayores o iguales a 30 para los tipos de tumores seleccionados (30
mutaciones representan una frecuencia de 1
por Mbp). Los histogramas anidados debajo de los gráficos circulares
muestran el porcentaje de pacientes sin genes
mutados de manera significativa (SMG, calculado por MutSig, q ≤ 0,1),
sin genes en el censo de cáncer (CGC) o
sin mutaciones en absoluto. Los datos de secuenciación de todos los
tumores pediátricos, el CCR y el carcinoma
hepatocelular se generaron del Centro de secuenciación del genoma
humano del Baylor College of Medicine. Los
datos de secuenciación de todos los demás tumores adultos proceden del
Centro de análisis de datos del genoma
de TCGA (https://confluence.broadinstitute.org/display/GDAC/Home). Los
datos para la LMA pediátrica, la LLA y
el tumores de Wilms se obtuvieron del proyecto TARGET
(http://www.targetproject.net/) (Figura modificada con
autorización de Wheeler DA. Genome Res. 2013 Jul;23(7):1054–1062.).
El patrón establecido desde la primera
evaluación exómica del cáncer colorrectal y de seno
demostró desde 2007 que un tercio de los genes están
mutados en más del 20% de los tumores. De estos, un
10% tienen mayor compromiso, y el resto generan una
menor carga genómica. Los proyectos a gran escala
como el TCGA y el ICGC han tenido como objetivo secuenciar más de 500
pacientes por cada tipo de tumor,
con la expectativa de recolectar una fracción considerable de los genes
mutados en un rango menor al 3%
(
Figura 3). Los genes alterados
en menor frecuencia
tienen gran importancia para la comprensión de la biología tumoral dado
que pueden presentar redundancia
mutacional en una vía específica de señalización. Esta
característica se hace evidente en el cáncer colorrectal
donde APC es el mayor impulsor de la vía canónica
WNT; sin embargo, otros 10 genes presentan cambios
deletéreos en el 1-15%, hallazgo que influye en la regulación positiva
de MYC y de múltiples factores de
transducción relacionados con PI3K (
Figura
4). Los
genes alterados con baja frecuencia revelan una complejidad adicional
en relación a la heterogeneidad clonal que favorece la resistencia a
medicamentos como
los inhibidores de tirosin-quinasa, hallazgo común en
los adenocarcinomas de pulmón EGFR (receptor para
el factor de crecimiento epidérmico) positivos.
Cuando se amplía la secuenciación del ADN incluyendo análisis del
número de copias, de expresión de
ARN, y perfiles epigéneticos, se modifica la información basal que se
obtiene. En general, la mayoría de
los perfiles mutacionales (como el que se muestra en
la Figura 3) identifican entre 15 y 20 genes mutados,
el análisis del número de copias agrega otros 20 genes
amplificados o con deleciones focales recurrentes, y los
perfiles epigenéticos y de expresión anormal refuerzan
los datos sobre las mutaciones somáticas agregando
usualmente algunos genes más. Según los resultados
obtenidos hasta ahora, parece probable que el repertorio de genes
implicados en cualquier tipo de cáncer sea
del orden de 50 a 100, en lugar de 500 a 1.000 como se
creía previamente.
Nuevas alteraciones genómicas de alta
y baja frecuencia
Desde el 2004 se han encontrado una multiplicidad de
alteraciones de baja frecuencia (menor del 20%) que
promueven la generación y sostenibilidad de neoplasias sólidas y
hematológicas. Los avances más notables
proporcionaron información sobre el papel de la remodelación de la
cromatina en la tumorigénesis. La isocitrato deshidrogenasa 1 y 2 (IDH1
y 2) fueron adiciones
significativas a la lista de impulsores del glioblastoma,
la LMA y el colangiocarcinoma (30-32). Ambas enzimas convierten el
isocitrato en α-cetoglutarato (αKG), un cofactor de las dioxigenasas
α-KG, incluidas
las ADN demetilasas de la familia TET, las histonas
demetilasas de la familia KDM y muchas otras proteínas (33). Cuando
IDH1 o 2 están mutados producen
2-oxiglutarato, un análogo estructural del α-KG que
actúa como potente inhibidor de enzimas dependientes de α-KG, las
metiltransferasas involucradas en la
metilación del ADN y la cromatina. Estos inhibidores
dan como resultado una modificación epigenética aberrante que se
transfiere a la desregulación de muchas
vías celulares.
La ADN demetilasa, DNMT3A, está mutada en el
22% de los pacientes con LMA, lo que sugirió que su
papel era importante para la regulación transcripcional a través de la
modificación epigenética del ADN
(34). Las mutaciones en este gen son clínicamente importantes en los
pacientes con LMA, ya que se asocian a reducción en la supervivencia
global. Desde su
descubrimiento inicial, las alteraciones en DNMT3A
se han relacionado con la transformación de casi todas las neoplasias
mieloides. De manera similar, las
alteraciones en el gen PBRM1 se documentaron en el
41% de los carcinomas renales de células claras, y este fue el primer
miembro del complejo para remodelado
de cromatina SWI/SNF que se encontró alterado en
cáncer (35). Desde entonces, las mutaciones en muchos de los otros
componentes de SWI/SNF se han
ido acumulando de manera constante en muestras de
pacientes con cáncer de ovario, hepatocelular, gástrico
y otros (36).
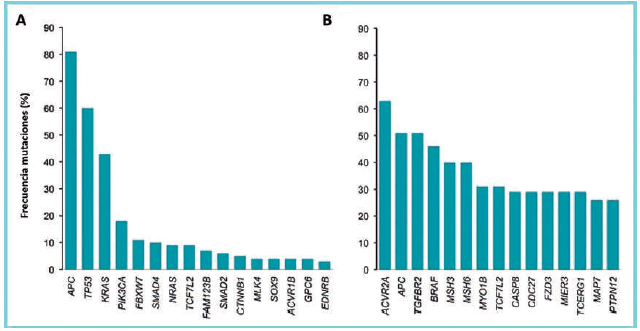
Figura 3. Genes mutados y principales vías desreguladas por
mutaciones somáticas en el carcinoma colorrectal
humano. Los pacientes se dividen en dos grupos según la tasa de
mutación. Todos los genes mostrados están
mutados significativamente con una tasa de descubrimiento <0,1. A.
Perfil mutacional en 193 pacientes con
inestabilidad microsomal; subgrupo con baja tasa mutacional (CRC
estabilidad de microsatélites). B. Perfil
mutacional en 29 pacientes con CRC positivo para inestabilidad
microsatélital, más 7 con mutación POLE (The
Cancer Genome Atlas Research Network).
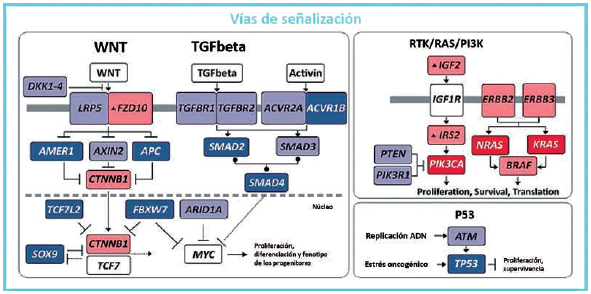
Figura 4. Principales vías desreguladas en carcinoma colorrectal.
Las alteraciones se definen por la presencia
de mutaciones somáticas, deleciones homocigotas, amplificaciones
focales de alto nivel y, en algunos casos,
por regulación de la expresión génica hacia arriba o hacia abajo. Todos
los genes de La señalización de WNT se
interrumpe por una o más mutaciones en el 93% de los pacientes. La
señalización de TGFbeta se interrumpe en
el 26% de los pacientes, y la de RTK/RAS/PI3K se limita en más del 80%
de los pacientes. En rojo se encuentran
los genes activados y en azul los inactivados. Los genes más claros no
presentan mutaciones significativas, pero
contribuyen con la alteración de la vía en algunos de los pacientes
(The Cancer Genome Atlas Research Network).
El gen para la proteína 1 asociada a BRCA (BAP1)
tiene una función enzimática que desubiquitina la
histona H2A y otras proteínas involucradas en la remodelación de la
cromatina. Presenta mutaciones
inactivantes en el 23% de los mesoteliomas (37) y en
los melanomas uveales, donde el 84% de los pacientes portadores tiene
alto riesgo de metástasis (38).
También se encuentra mutado en el 15% de los carcinomas renales de
células claras, en los que está anticorrelacionado con las mutaciones
más frecuentes de
PBRM1, mencionadas anteriormente. Una mutación
inactivante en BAP1 define una subclase molecular de
tumores agresivos de alto grado.
En el otro extremo del perfil mutacional están los genes del cáncer que
contribuyen al 10% o menos de los
tumores. Quizás los más interesantes en el ámbito de la
baja carga y frecuencia son aquellos relacionados con
el procesamiento del ARN. Descubiertos por primera
vez en el síndrome mielodisplásico, los genes U2AF1,
U2AF2, SF3B1 y SRSF2 (39), están involucrados en el
reconocimiento del aceptor de empalme en la maquinaria del ARN, mutaron
acumulativamente en más del
50% de los pacientes.
Heterogeneidad tumoral
La naturaleza estocástica del cáncer refuerza la noción de que el
desarrollo y progresión del cáncer no
sigue un curso lineal fijo, sino más bien, una desestabilización
integrada de múltiples procesos celulares
clave. Incluso después de la transformación maligna,
el cáncer permanece dinámico y continúa evolucionando para generar un
ecosistema heterogéneo con
elementos formes que albergan formas moleculares
distintas, con niveles diferenciales de sensibilidad a
las terapias. Esta heterogeneidad puede resultar de
cambios genéticos, transcriptómicos, epigenéticos
y/o fenotípicos. A nivel poblacional, la heterogeneidad tumoral puede
dividirse en intertumoral e intratumoral; la heterogeneidad
intertumoral se refiere a
los cambios entre pacientes que tienen tumores del
mismo linaje histológico y se relacionan con variaciones germinales, en
el perfil de mutaciones somáticas,
y factores ambientales. Por otra parte, la heterogeneidad intratumoral
puede manifestarse como cambios a
nivel espacial, que describen la distribución desigual
de subpoblaciones tumorales genéticamente diversas
en diferentes sitios de la enfermedad o dentro de una
sola enfermedad, sitio o tumor, y como heterogeneidad temporal, un
término aplicado a la diversidad
genética de un tumor individual a lo largo del tiempo
(procesos endógenos (como la replicación del ADN
y/o errores de reparación, estrés oxidativo) (40). La
Figura 5 describe gráficamente
los diferentes tipos de
heterogeneidad tumoral.
En conjunto, las principales causas de heterogeneidad
tumoral son la inestabilidad genómica y la evolución
y selección clonal. La primera, puede variar en magnitud, desde las
sustituciones de una sola base hasta
duplicaciones completas del genoma. Tal inestabilidad
puede resultar de la exposición a mutágenos exógenos
(como radiación UV o humo por combustión de tabaco) y aberraciones en
los procesos endógenos (como la
replicación del ADN y/o errores de reparación, estrés
oxidativo) (40). Por ejemplo, la inestabilidad microsatelital,
relacionada a deficiencias en la reparación del
ADN (MMR), impulsa la transformación neoplásica
y aumenta sustancialmente la carga de mutaciones somáticas de un
subconjunto de cáncer colo rectal (CCR)
y otros cánceres con este fenotipo distintivo. Estudios
que involucran el análisis del genoma completo han
permitido la identificación de firmas genéticas características
asociadas con algunos de estos procesos mutagénicos que favorecen la
heterogeneidad. Por ejemplo,
las neoplasias de pulmón ligadas a la exposición al
humo por combustión del tabaco están enriquecidos
con transversiones C>A y, de manera similar, los CCR
deficientes en MMR son más propenso a las transiciones C>T. Además,
aunque no contribuye a la inestabilidad genómica basal, la exposición a
la quimioterapia
también puede aumentar el espectro mutacional un tumor generando
deregulación genómica. Según ciertas
teorías, la tumorigénesis es dependiente de una elevada tasa de
mutaciones espontáneas (41). Esta hipótesis
no ha sido probada definitivamente y puede que no
sea aplicable a todos los cánceres, aunque los datos de
múltiples estudios han demostrado que los cánceres
a menudo tienen procesos homeostáticos endógenos
con el fin de aumentar la carga mutacional global. Por
ejemplo, la deaminación de citosinas del ADN, resultante de la
regulación al alza del mismo a través de la
dC → dU-enzima de edición (APOBEC3B) contribuye con la mutagénesis. La
firma mutacional de APOBEC, que se caracteriza por la presencia de
mutaciones
C>T y C>G en los sitios TpC, está particularmente
enriquecida en las etapas avanzadas del desarrollo del
tumor, y se hace más prevalente después de la exposición a la
quimioterapia. Los altos niveles de expresión
de APOBEC3B presagian un peor resultado entre pacientes con ciertas
neoplasias sólidas; por ejemplo, un
estudio sobre el análisis de 1.500 muestras de cáncer
de seno reveló una asociación entre los altos niveles de
expresión de APOBEC3B y una menor supervivencia
libre de enfermedad y global. La inhibición de estas
enzimas podría reducir la inestabilidad genética y mejorar así los
resultados de los pacientes, en particular,
en tumores cerebrales. La inestabilidad genómica también puede conducir
a inestabilidad cromosómica por
errores de segregación que ocurren durante la división
celular, alterando el equilibrio entre la activación de
los oncogenes y los genes supresores de tumor (42).
La evolución clonal también puede darse por desequilibrio en el número
de copias y por la pérdida
uniforme de segmentos cromosómicos que albergan
segmentos génicos específicos.
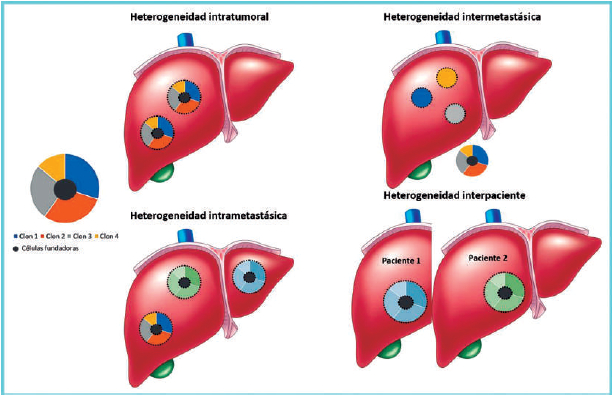
Figura 5. Manifestaciones gráficas de la heterogeneidad tumoral
Estos hallazgos permiten documentar variaciones intratumorales de orden
subclonal que capacitan a las células en derivados más
competitivos. Se han propuesto varios modelos para
explicar cómo la diversidad clonal se genera y mantiene, aunque la
mayoría continúan utilizando el marco de selección propuesto por Peter
Nowell en 1976,
basado en la hipótesis de que la iniciación del cáncer
ocurre gracias a la ventaja de crecimiento selectivo que
conduce a la rápida proliferación. Posteriormente, la
inestabilidad genómica de la población de tumores en
expansión crea más diversidad genética que está sujeta a presiones de
selección evolutiva, lo que resulta
en la emergencia secuencial de células cada vez más
anormales genéticamente. La evolución lineal de la
enfermedad permite la adquisición sucesiva de mutaciones que
proporcionan superioridad sobre el ancestro común. Alternativamente, la
evolución ramificada
denota la aparición y propagación divergente de múltiples poblaciones
de células tumorales subclonales que
comparten un ancestro común. La evolución ramificada permite un mayor
nivel de oportunidad para crear
un tumor heterogéneo. La mayoría de las neoplasias
sólidas optan por un patrón ramificado de evolución,
mientras las hematológicas invocan la secuencia lineal. Curiosamente,
estos supuestos han sido desafiados por la necesidad de cooperación
celular entre
distintos subclones para la propagación tumoral (43).
Vías de señalización
A pesar de la inmensa complejidad de los genomas
tumorales, progresivamente hemos adquirido la capacidad de controlar
algunos de los genes conductores,
como lo demuestran las respuestas en pacientes con
alteraciones particulares en EGFR (receptor para el
factor de crecimiento epidérmico), ALK (quinasa del
linfoma anaplásico) y BRAF (homólogo B del oncogén viral del sarcoma
murino v-Raf), entre otros (44).
Aunque transitorias, significan que la interferencia con
un solo producto génico mutado es suficiente para detener temporalmente
la evolución de la enfermedad.
Hay dos conceptos esenciales al momento de considerar esta información
para sustentar el uso de la oncología de precisión. Primero, el 99% de
las alteraciones
genéticas del cáncer (incluidas las mutaciones puntuales, alteraciones
en el número de copias, translocaciones y cambios epigenéticos
distribuidos a lo largo del
genoma, no solo en las regiones codificantes), como se
expuso previamente, son irrelevantes para la neoplasia. Son simplemente
cambios pasajeros que marcan
el tiempo que ha transcurrido entre las sucesivas expansiones clonales.
Sin embargo, las células normales
están programadas para ejecutar la muerte celular en
respuesta a tales alteraciones, quizás como mecanismo
protector contra el cáncer. Por el contrario, las células
tumorales han evolucionado para tolerar la complejidad estructural del
genoma mediante la adquisición de
cambios en genes como TP53 (45). Por tanto, la complejidad genómica es,
en parte, el resultado del cáncer,
más que la causa.
También hay cierto orden en el caos de las neoplasias.
Las mutaciones en los genes conductores causan una
ventaja de crecimiento selectivo, ya sea directa o indirectamente, y
siempre a través de un número limitado
de vías de señalización celular (todos se pueden clasificar en 12
vías). El descubrimiento de los componentes moleculares de estas vías
es uno de los mayores
logros de la investigación biomédica, un tributo a los
investigadores que trabajan en campos que abarcan la
bioquímica, la biología celular, y el cáncer. Estas vías
pueden organizarse en sí mismas en tres procesos celulares centrales
incluyendo el devenir celular, la supervivencia, y el mantenimiento del
genoma. Numerosos
estudios han demostrado la relación opuesta entre la
división y diferenciación celular. Las células activas
que son responsables de poblar los tejidos normales no
se diferencian, y viceversa. La ventaja de crecimiento
selectivo esta favorecida por la actividad de vías que incluyen APC, HH
y NOTCH, que son bien conocidas por controlar el destino celular. Los
genes que
codifican las enzimas modificadoras de la cromatina
también pueden incluirse en esta categoría (46). En el
desarrollo normal, el cambio hereditario de la división
a la diferenciación no está determinado por la mutación, como ocurre en
el cáncer, sino por las alteraciones epigenéticas que afectan al ADN y
las proteínas de
la cromatina.
Aunque las células neoplásicas se dividen de forma
anormal debido a sus alteraciones autónomas, las
células estromales que las rodean son perfectamente
normales y no siguen el mismo ritmo. La ramificación
más obvia de esta asimetría es la vasculatura anormal
de los tumores (47). A diferencia de la red bien ordenada de arterias,
venas y linfáticos que controlan las
concentraciones de nutrientes en los tejidos normales,
el sistema vascular en los cánceres es tortuoso y carece de uniformidad
estructural. Las células normales
están siempre a menos de 100 μm de un capilar, pero
esto no es cierto para las células tumorales (48). Como
resultado, una célula neoplásica que adquiera una mutación que le
permita proliferar bajo concentraciones
limitantes de nutrientes y oxígeno tendrá una ventaja
de crecimiento selectivo, prosperando en entornos claramente hostiles
que exigen adaptación. Las mutaciones que sustentan la supervivencia a
través de vías de
señalización están relacionadas con los genes
EGFR,
HER2, FGFR2, PDGFR, TGFbR2, MET, KIT, RAS,
RAF, PIK3CA y PTEN. Algunos de estos genes codifican receptores
para los propios factores de crecimiento, mientras que otros transmiten
la señal directamente al interior de la célula, estimulando el
crecimiento
cuando se activa. Por ejemplo, las mutaciones en los
genes
KRAS o BRAF confieren a
las células anormales
la capacidad de crecer en concentraciones de glucosa
inferiores a las requeridas para el crecimiento de células normales
(49). La progresión a través del ciclo
celular (y su antítesis, la apoptosis) puede controlarse directamente
mediante metabolitos intracelulares,
y los genes impulsores que regulan directamente el
ciclo celular o la apoptosis, como
CDKN2A,
MYC y
BCL2, a menudo están mutados en paralelo. Otro gen
cuyas mutaciones potencian la supervivencia celular
es
VHL, cuyo producto
estimula la angiogénesis mediante la secreción del factor de
crecimiento vascular
endotelial (
VEGF) y con el
relacionado con la hipoxia
(
HIF1). Las vías de conducción
intracelular relacionadas con el mantenimiento del genoma son
esenciales
para mantener el microambiente interno de la célula
tumoral y su estabilidad genómica. Por tanto, no es de
extrañar que mutaciones en los genes que mantienen o
anulan puntos de control, como
TP53
y ATM, favorecen la proliferación (50).
Conclusiones
Aunque la secuenciación del genoma del cáncer es
un esfuerzo relativamente nuevo, ha tenido un enorme impacto en la
atención clínica de los pacientes con
diversas neoplasias. El reconocimiento de que ciertos
tumores contienen mutaciones activadoras en genes
conductores que codifican proteínas quinasas ha llevado al desarrollo
de fármacos inhibidores específicos
(51). Ejemplos representativos de este tipo de medicina basada en el
genoma incluyen el uso de inhibidores
del
EGFR, BRAF y ALK antes
mencionados. Solo una
fracción de los pacientes con cáncer de pulmón tienen
mutaciones en el gen
EGFR o
translocaciones de
ALK,
y responderán a los medicamentos blanco dirigidos.
Sin embargo, la genómica ha identificado 16 blancos
potenciales en desarrollo para la misma enfermedad,
alcanzando diferentes líneas para cada ámbito (52).
Un segundo tipo de medicina basada en la genómica
se centra en los efectos secundarios y el metabolismo
de los agentes terapéuticos, más que en las alteraciones genéticas a
las que se dirigen. En la actualidad,
la dosis de los medicamentos contra el cáncer se basa
en el área de superficie, pero la proporción terapéutica
o biológica efectiva depende de la interacción génica.
De manera óptima, la evaluación del genoma permitirá realizar
mediciones farmacocinéticas de las concentraciones biológicamente
activas en cada paciente
(53). El costo adicional de tales análisis sería menor en
comparación con los costos exorbitantes de los medicamentos, que se
estima en US$200.000 - US$300.000
por año de calidad de vida ganado con calidad.
Referencias
1. Dulbecco R. A turning point in
cancer research: sequencing the human genome. Science. 1986; 231(4742):
1055-6.
2. Robertson M. The proper study of
mankind. Nature.
1986; 322(6074):11.
3. Futreal PA, Coin L, Marshall M, et
al. A census of human
cancer genes. Nat Rev Cancer. 2004; 4(3):177-83.
4. Vogelstein B, Papadopoulos N,
Velculescu VE, et al.
Cancer genome landscapes. Science. 2013; 339(6127):
1546–1558.
5. Herceg Z, Hainaut P. Genetic and
epigenetic alterations
as biomarkers for cancer detection, diagnosis and prognosis. Mol Oncol.
2007;1(1):26–41.
6. Sadikovic B, Al-Romaih K, Squire
JA, Zielenska M. Cause
and consequences of genetic and epigenetic alterations
in human cancer. Curr Genomics.2008;9(6):394–408.
7. Stankiewicz P, Lupski JR.
Structural variation in the human genome and its role in disease. Annu
Rev Med.
2010;61:437–455.
8. Talpaz M, Silver RT, Druker BJ,
Goldman JM, Gambacorti-Passerini G, Guilhot BJ et al. Imatinib induces
durable hemato-logic and cytogenetic responses in patients
with accelerated phase chronic myeloid leukemia: results of a phase 2
study. Blood. 2002;99(6):1928–1937.
9. Eckschlager T, Pilat D, Kodet R,
et al. DNA ploidy in neuroblastoma. Neo-plasma. 1996;43(1):23–26.
10. Mandahl N, Gustafson P, Mertens
F, et al. Cytogenetic
aberrations and their prognostic impact in chondrosarcoma. Genes
Chromosomes Cancer. 2002;33(2):188–200.
11. Orr LC, Fleitz J, McGavran L, et
al. Cytogenetics in
pediatric low-grade astrocytomas. Med Pediatr Oncol.
2002;38(3):173–177.
12. Biswas S, Rao CM. Epigenetics in
cancer: fundamentals
and beyond. Pharmacol Ther. 2017;173:118–134.
13. Chen ZX, Riggs AD. DNA
methylation and demethylation
in mammals. J Biol Chem. 2011;286(21):18347–18353.
14. Chakravarthi BV, Nepal S,
Varambally S. Genomic
and epigenomic alterations in cancer. Am J Pathol.
2016;186(7):1724–1735.
15. Weisenberger DJ. Characterizing
DNA methylation alterations from The Cancer Genome Atlas. J Clin
Invest.
2014;124(1):17–23.
16. Zhao R, Choi BY, Lee MH, et al.
Implications of genetic
and epigenetic alterations of CDKN2A (p16(INK4a)) in
cancer. EBioMedicine. 2016;8:30–39.
17. Reddy KB. MicroRNA (miRNA) in
cancer. Cancer Cell
Int. 2015;15:38.
18. Chakraborty C, Sharma AR, Sharma
G, et al. The novel strategies for next-generation cancer treatment:
miRNA combined with chemotherapeutic agents for the
treatment of cancer. Oncotarget. 2018;9(11):10164–
10174.
19. Dizdaroglu M. Oxidatively induced
DNA damage
and its repair in cancer. Mutat Res Rev Mutat Res.
2015;763:212–245.
20. Feinberg AP, Koldobskiy MA,
Gondor A. Epigenetic modulators, modifiers and mediators in cancer
aetiology
and progression. Nat Rev Genet. 2016;17(5):284–299.
21. Wallace SS, Murphy DL, Sweasy JB.
Base excision repair and cancer. Cancer Lett. 2012;327(1–2):73–89.
22. Kim YJ, Wilson DM, 3rd. Overview
of base excision repair biochemistry. Curr Mol Pharmacol.
2012;5(1):3–13.
23. Le DT, Durham JN, Smith KN, et
al. Mismatch repair deficiency predicts response of solid tumors to
PD-1 blockade. Science. 2017;357(6349):409–413.
24. Li SKH, Martin A. Mismatch repair
and colon cancer:
mechanisms and therapies explored. Trends Mol Med.
2016;22(4):274–289.
25. Helleday T, Eshtad S, Nik-Zainal
S. Mechanisms underlying mutational signatures in human cancers. Nat
Rev Genet. 2014;15(9):585–598.
26. Aparicio T, Baer R, Gautier J.
DNA double-strand break
repair pathway choice and cancer. DNA Repair (Amst).
2014;19:169–175.
27. Chang HHY, Pannunzio NR, Adachi
N, Lieber MR. Nonhomologous DNA end joining and alternative pathways
to double-strand break repair. Nat Rev Mol Cell Biol.
2017;18(8):495–506.
28. Katsanis SH, Katsanis N.
Molecular genetic testing
and the future of clinical genomics. Nat Rev Genet.
2013;14(6):415–426.
29. Futreal PA, Coin L, Marshall M,
et al. A census of human
cancer genes. Nat Rev Cancer. 2004 ; 4(3):177-83.
30. Tokheim CJ, Papadopoulos N,
Kinzler KW, et al. Evaluating the evaluation of cancer driver genes.
Proc Natl
Acad Sci U S A. 2016;113(50):14330-14335..
31. Parsons DW, Jones S, Zhang X, et
al. An integrated
genomic analysis of human glioblastoma multiforme.
Science. 2008; 321(5897):1807-12.
32. Hou AH, Tien HF. Genomic
landscape in acute myeloid
leukemia and its implications in risk classification and
targeted therapies. J Biomed Sci. 202021;27(1):81.
33. Chen G, Cai Z, Dong X, et al.
Genomic and Transcriptomic Landscape of Tumor Clonal Evolution in
Cholangiocarcinoma. Front Genet. 2020;11:195.
34. Yang H, Ye D, Guan KL, et al.
IDH1 and IDH2 mutations
in tumorigenesis: mechanistic insights and clinical perspectives. Clin
Cancer Res. 2012;18(20):5562-71.
35. Ley TJ, Ding L, Walter MJ, et al.
DNMT3A mutations in acute myeloid leukemia. N Engl J Med.
2010;363(25):2424-33.
36. Varela I, Tarpey P, Raine K, et
al. Exome sequencing identifies frequent mutation of the SWI/SNF
complex gene PBRM1 in renal carcinoma. Nature.
2011;469(7331):539-42.
37. Shain AH, Pollack JR. The
spectrum of SWI/SNF mutations, ubiquitous in human cancers. PLoS One.
2013;
8(1):e55119.
38. Kobrinski DA, Yang H, Kittaneh M.
BAP1: role in carcinogenesis and clinical implications. Transl Lung
Cancer
Res. 2020;9(Suppl 1):S60-S66.
39. Li Y, Shi J, Yang J, et al. Uveal
melanoma: progress in
molecular biology and therapeutics. Ther Adv Med Oncol. 2020 Oct
22;12:1758835920965852.
40. Yoshida K, Sanada M, Shiraishi Y,
Nowak D, Nagata Y, Yamamoto R et al. Frequent pathway mutations of
splicing machinery in myelodysplasia. Nature.
2011;478(7367):64-9.
41. Dagogo-Jack I, Shaw AT. Tumour
heterogeneity and
resistance to cancer therapies. Nat Rev Clin Oncol.
2018;15(2):81-94.
42. Burrell RA, McGranahan N, Bartek
J, Swanton C. The
causes and consequences of genetic heterogeneity in
cancer evolution. Nature. 2013;501(7467):338-45.
43. Turner NC, Reis-Filho JS. Genetic
heterogeneity and
cancer drug resistance. Lancet Oncol. 2012 ;13(4):e178-
85. Mar 30.
44. Venkatesan S, Swanton C, Taylor
BS, Costello JF.
Treatment-Induced Mutagenesis and Selective Pressures Sculpt Cancer
Evolution. Cold Spring Harb Perspect
Med. 2017;7(8):a026617.
45. Zhang Y, Ma Y, Li Y, et al.
Comparative analysis of cooccurring mutations of specific tumor
suppressor genes
in lung adenocarcinoma between Asian and Caucasian
populations. J Cancer Res Clin Oncol. 2019;145(3):747-
757.
46. Ljungman M, Lane DP.
Transcription - guarding the
genome by sensing DNA damage. Nat Rev Cancer.
2004;4(9):727-37.
47. Perrimon N, Pitsouli C, Shilo BZ.
Signaling mechanisms
controlling cell fate and embryonic patterning. Cold
Spring Harb Perspect Biol. 2012;4(8):a005975.
48. Baish JW, Stylianopoulos T,
Lanning RM, Kamoun WS,
Fukumura D, Munn L et al. Scaling rules for diffusive
drug delivery in tumor and normal tissues. Proc Natl
Acad Sci U S A. 2011; 108(5):1799-803.
49. Turner N, Grose R. Fibroblast
growth factor signalling:
from development to cancer. Nat Rev Cancer. 2010;
10(2):116-29.
50. Yun J, Rago C, Cheong I, et al.
Glucose deprivation contributes to the development of KRAS pathway
mutations
in tumor cells. Science. 2009; 325(5947):1555-9.
51. Medema RH, Macůrek L. Checkpoint
control and cancer. Oncogene. 2012; 31(21):2601-13.
52. Bertier G, Carrot-Zhang J,
Ragoussis V, Joly Y. Integrating precision cancer medicine into
healthcare—policy,
practice, and research challenges. Genome Med. 2016;
8(1):108.
53. Malone ER, Oliva M, Sabatini PJB,
Stockley TL, Siu LL.
Molecular profiling for precision cancer therapies. Genome Med.
2020;12(1): 8.
54. McLeod HL. Cancer
pharmacogenomics: early promise, but concerted effort needed. Science.
2013;
339(6127):1563-6.
Recibido:
Noviembre 26, 2020
Aprobado: Noviembre 26, 2020
Correspondencia:
Andrés F. Cardona
cardona@clinicadelcountry.com