CARACTERIZACIÓN DE LA EPILEPSIA BENIGNA DE LA INFANCIA CON PUNTAS CENTROTEMPORALES* ESTUDIO DE CORTE TRANSVERSAL REALIZADO EN NIÑOS ATENDIDOS EN DOS LABORATORIOS DE REFERENCIA DE LA CIUDAD MEDELLÍN (2011-2016)
Yuly Mildred Bayona Ovalles1, Diana Cornejo Sánchez2, Isabel Camacho Ordoñez3, José William Cornejo Ochoa4, Rodrigo Andrés Solarte Mila5
RESUMEN
Introducción: La epilepsia benigna de la infancia con puntas centro-temporales es el síndrome epiléptico focal más común en pediatría. No hay datos sistematizados en Colombia sobre esta enfermedad. Objetivo: Describir las características clínicas, electroencefalográficas y familiares de este síndrome. Materiales y métodos: Estudio descriptivo, retrospectivo, de corte transversal, con análisis exploratorio. Se incluyeron pacientes <18 años que consultaron a 2 laboratorios de electroencefalografía (EEG) en Medellín, Colombia, de 2011 a 2016, que cumplieron criterios diagnósticos de epilepsia rolándica, con EEG que mostraba puntas centro-temporales con máxima negatividad en los electrodos centro-temporales (C3, C4 y T3,T4), con activación en sueño. Se calcularon las frecuencias y proporciones para variables cualitativas y promedios y desviaciones estándar (DE) para las cuantitativas. En el análisis exploratorio se utilizaron las pruebas de chi cuadrado, test exacto de Fisher y test Shapiro –Wilk. Resultados: Se incluyeron 44 pacientes, 46% mujeres, 54% hombres. La edad promedio de inicio de la epilepsia fue 6,6 años (DE: 2,3). Las manifestaciones clínicas más frecuentes de esta epilepsia fueron: Sialorrea 27% y sonidos faríngeos 27%. Las crisis se presentaron durante el sueño en 43%, no muy diferente del porcentaje en vigilia, 46%. El 76% había presentado máximo 4 crisis. No hubo diferencias respecto a la lateralidad de la actividad epileptiforme en el EEG. La fase del sueño en la que más se presentó la actividad epileptiforme fue N2. En el 57%, la actividad se presentaba en salvas. Solo el 20% presentó actividad extra centro-temporal. El 32% de los pacientes tenía antecedente familiar de epilepsia. Las comorbilidades más frecuentes fueron bajo rendimiento escolar 34%, trastorno del lenguaje 25% y TDAH 23%. El 61% de los pacientes recibía tratamiento farmacológico y el medicamento más usado fue la carbamazepina en 25%. En el análisis exploratorio se encontró que la frecuencia de los complejos POL era más baja: 3,3 Hz, en los pacientes con trastorno del lenguaje que en los pacientes sin esta comorbilidad: 3,6 Hz, (p=0.02). Conclusión: Aunque el tamaño de la muestra de este estudio no es grande, tiene el valor de ser la primera descripción clínica, electroencefalográfica y familiar de la epilepsia rolándica en Colombia. Las características clínicas son similares a lo descrito en otras series, pero con mayor proporción de generalización de las crisis. La frecuencia en Hz de los complejos POL estuvo por encima de lo previamente informado. El antecedente familiar de epilepsia fue 3 veces más alto en los pacientes de este estudio, lo que podría sugerir que existen factores genéticos y/o ambientales comunes entre este síndrome y otros tipos de epilepsia en nuestra población. Las comorbilidades más frecuentes fueron bajo rendimiento escolar, TDAH y trastorno del lenguaje, los cuales se deben buscar activamente en estos pacientes. La frecuencia en Hz de los complejos POL podría ser un marcador de pronóstico respecto al lenguaje en pacientes con epilepsia rolándica.
Palabras clave: Epilepsia rolándica, epilepsia benigna de la infancia con puntas centro-temporales, electroencefalograma.
_______________________
* Esta investigación participó en el Premio Nacional de Epilepsia Margaret Merz de Fandiño, 2017.
1. Pediatra, Neuróloga Infantil, Universidad de Antioquia.
2. Estudiante de Doctorado Ciencias Básicas Biomédicas, Grupo de investigación Mapeo Genético, Universidad de Antioquia.
3. Pediatra, Neuróloga infantil. Profesora asociada del Departamento de Pediatría, Facultad de Medicina, Universidad de Antioquia.
4. Neurólogo infantil, Magíster en Epidemiología. Profesor Titular del Departamento de Pediatría, Grupo de Investigación Pediaciencias, Facultad de Medicina, Universidad de Antioquia.
5. Neurólogo, Epileptólogo. Profesor asociado del Departamento de Pediatría, Facultad de Medicina, Universidad de Antioquia.
________________________
CHARACTERIZATION OF CHILDHOOD BENIGN EPILEPSY WITH CENTROTEMPORAL SPIKES. CROSS-SECTIONAL STUDY DONE IN CHILDREN SEEN IN REFERENCE LABORATORIES IN THE CITY OF MEDELLIN (2011-2016)
ABSTRACT
Introduction: Benign epilepsy with centro-temporal spikes (BECTS) is the most frequent focal epileptic syndrome in childhood. There are no systematic data about it at Colombia. Our objective was to describe the clinical, electroencephalographic and familiar characteristics of this syndrome. Materials and methods: Retrospective, descriptive, cross- sectional study, with exploratory analysis. Patients admitted were children under 18 years of age, who had attended one of two selected electroencephalography laboratories at Medellín, Colombia, from 2011 to
2016, and filled diagnostic criteria for rolandic epilepsy, EEG with centro-temporal spikes, maximal electronegativity at C3, C4 and T3, T4 electrodes, and activation during sleep. Frequencies and proportions were calculated for qualitative variables, averages and standard deviation for quantitative variables. Chi square, Fisher test and Shapiro-Wilk test were used for exploratory analysis. Results: 44 patients were included, 46% women and 54% men. Average age when epilepsy started was 6.6 years old (SD: 2.3). Most frequent symptoms were: Hypersalivation and oropharyngeal sounds with 27% each. Seizures presented during sleep in 43% with a similar proportion during awake. 76% had presented 4 seizures top. There were no differences in the side of epileptiform activity in EEG. N2 was the sleep phase with more epileptiform activity. In
57% of cases the epileptiform activity presented in clusters. Only 20% had extra centrotemporal activity. There was familiar history of epilepsy in 32%. The most frequent comorbidities were poor school performance: 34%, speech disorder: 25% and ADHD: 23%. 61% of patients were on treatment, and the most frequent medication was carbamazepine: 25%. The exploratory analysis showed that the frequency of spike and slow wave complexes were slower: 3.3 Hz, in patients with speech disorder than in patients without it: 3.6 Hz (p=0.02). Conclusion: This is the first clinical, electroencephalographic and familiar description of rolandic epilepsy in our country. Clinical presentation was similar to other reports, but our patients had more generalization of seizures. The frequency of spike and slow wave complexes (Hz) were in general above of previous reports. Family history of epilepsy was three times higher in patients of this study, suggesting that there are some common genetic and environmental factors between this syndrome and other types of epilepsy in our population. The most frequent comorbidities were poor school performance, speech disorder, and ADHD. This should be carefully evaluated in patients with this syndrome. The frequency of spike and slow wave complexes (Hz) could be a marker of speech prognosis in patients with rolandic epilepsy.
Keywords: Rolandic epilepsy, benign epilepsy with centro-temporal spikes, electroencephalography.
INTRODUCCIÓN
La epilepsia benigna de la infancia con puntas centrotemporales o epilepsia rolándica es el síndrome epiléptico focal más común en la edad pediátrica, constituye el 23-25% de los síndromes epilépticos diagnosticados. Se caracteriza por crisis focales sensitivas y motoras involucrando frecuentemente a los músculos de la laringe, faringe y lengua, interfiriendo en el habla hasta llegar a la anartria, acompañándose de sialorrea y que pueden evolucionar a crisis tónico clónica bilateral. El síndrome tiende a desaparecer en la adolescencia por lo que ha sido considerado de naturaleza benigna. El nombre aceptado por la ILAE (International League Against Epilepsy) es epilepsia benigna de la infancia con puntas centro-temporales (BCECTS, por sus siglas en inglés), reflejando de manera más exacta las características clínicas y electroencefalográficas, pero el término epilepsia rolándica es su sinónimo ampliamente conocido (1-4).
La ILAE define BCETCS como un síndrome con crisis focales sin alteración de conciencia, de corta duración, localización hemifacial, frecuentemente asociadas a sintomatología somatosensorial ipsilateral, que pueden evolucionar a crisis tónico clónica bilateral. Las crisis se presentan en su mayoría durante el sueño. Su inicio ocurre entre los 3 a 13 años de edad, con un pico de incidencia a los 9-10 años, y su resolución se da antes de los 15-16 años de edad. En el 75% de los pacientes, el inicio se da entre los 7-10 años. Su incidencia es de 4,7-20/100.000 niños de 0 a 15 años de edad, con una prevalencia de 15-25% en niños de 1 a 15 años con epilepsia, y una predominancia masculina de 1,5:1 (5,6).
Este tipo de epilepsia se ha asociado a discalculia, dificultades en el lenguaje expresivo y receptivo (7,8), en el aprendizaje, dispraxia orolingual y visoespacial, dislexia y alteración de funciones ejecutivas (9). Las puntas centro- temporales son la característica del EEG de la epilepsia rolándica, pero es de aclarar que no todos los niños que presentan puntas centrotemporales desarrollan las características clínicas de la enfermedad (10).
La morfología de las ondas en el EEG es un factor determinante para definir la epilepsia rolándica, ya que se aprecian ondas difásicas o trifásicas de alto voltaje, de gran amplitud (hasta 200-300 uV) y con una duración promedio de 70-80 ms.
Estas ondas se presentan en regiones centrotemporales, de manera aislada o en brotes, seguidas de manera inconstante por ondas lentas, que están presentes durante la vigilia y el sueño NREM, con un ritmo de fondo normal en el EEG. El dipolo típico es horizontal, presenta máxima negatividad en regiones centrales y medio-temporales, con positividad en regiones frontales, pero con menor frecuencia pueden verse pacientes con solo negatividad centro temporal (11).
No existe hasta el momento correlación entre la gravedad de la epilepsia y el trazado electroencefalográfico (12-15).
Las teorías aceptadas sobre la etiología de la epilepsia rolándica apuntan a una patogénesis multifactorial que lleva a una alteración de la maduración del cerebro, siendo el factor genético el más estudiado (16).
El cromosoma 15 se ha destacado por su relación con BCECTS, específicamente 15q14 (que se encuentra relacionado también con la epilepsia mioclónica juvenil). Allí se encuentra localizado el gen que codifica la subunidad α7 del receptor nicotínico de acetilcolina y también, al parecer, el gen de un cotransportador del cloro (KCC4), por lo cual ambos genes están actualmente en estudio para esclarecer si mutaciones de éstos son causantes de las alteraciones observadas en BCECTS (17).
La decisión de iniciar o no un tratamiento anticonvulsivo sigue controvertida, dada la poca frecuencia de crisis en este síndrome y la tendencia natural del cuadro a resolución espontánea en la adolescencia.
Cuando se decide dar tratamiento, clásicamente se ha descrito que el medicamento de elección es la carbamazepina, aunque la efectividad de otros medicamentos como oxcarbazepina, fenobarbital y ácido valproico es similar. En algunos casos la carbamazepina puede producir trastornos del aprendizaje o empeoramiento de las crisis, por lo que se recomienda considerar otras opciones a la hora de iniciar tratamiento. La administración de clobazam nocturno es una buena opción en pacientes con crisis de predominio en sueño. Otras opciones útiles son el levetiracetam y lamotrigina. En evoluciones atípicas, el clonazepam ha mostrado ser útil, solo o en combinación (18).
Siendo esta una entidad tan frecuente en la infancia, es muy importante conocer qué tanto lo es en nuestra población (perfil clínico, electro-encefalográfico y cognitivo) y cuál es el comportamiento familiar de la misma, en busca de establecer diferencias con lo informado en otras series, considerando que no hay estudios al respecto en nuestro medio. Una vez obtenida esta información será posible avanzar en la búsqueda de factores etiológicos, principalmente genéticos en nuestra población y orientar futuras intervenciones para su diagnóstico y terapia integral.
MATERIALES Y MÉTODOS
Se realizó un estudio descriptivo, retrospectivo, de corte transversal, con análisis exploratorio. Se incluyeron todos los pacientes de la bases de datos del laboratorio CEC-LAB de la IPS Universitaria de Medellín y Vida y Cerebro IPS, menores de 18 años, de 2011 a 2016, que tenían diagnóstico de epilepsia rolándica, con EEG que mostrara puntas centrotemporales con máxima negatividad en los electrodos centrotemporales (C3, C4 y T3, T4) y que se activaban en sueño. Se debía disponer de los registros clínicos y EEG de los pacientes para que ingresara al estudio.
Se excluyeron pacientes que presentaran pérdida de grafoelementos de sueño en el EEG o actividad irritativa en EEG que sugiera compromiso estructural. Además aquellos con enfermedades neurodegenerativas, parálisis cerebral, discapacidad cognitiva.
Procedimiento de recolección de datos y procesamiento de la información
Se realizó una búsqueda en las bases de datos disponibles en los laboratorios CECLAB y Vida y Cerebro IPS, y se seleccionaron aquellos con diagnóstico de epilepsia rolándica según lo descrito previamente. Se evaluaron los registros correspondientes, verificando el cumplimiento de los criterios de inclusión y exclusión.
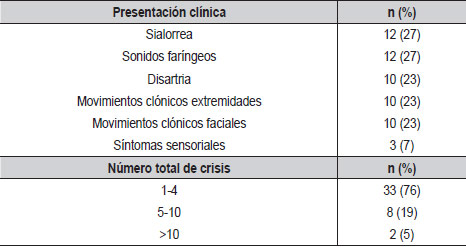
Se diligenció el instrumento de recolección de datos, extrayendo la siguiente información: Edad de inicio de epilepsia, sexo, lugar de nacimiento. Variables clínicas: Crisis en sueño, crisis en vigilia, crisis en sueño y vigilia, sialorrea, síntomas sensoriales, disartria, sonidos faríngeos, movimientos clónicos faciales, movimientos clónicos de extremidades, lateralidad de la crisis, generalización secundaria de la crisis, aviso de inicio de crisis, número de crisis hasta el momento de la evaluación y crisis prolongada.
Variables electroencefalográficas
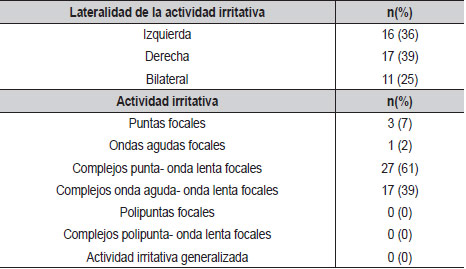
Lateralidad de las puntas centrotemporales, presencia de puntas focales, ondas agudas focales, complejos punta onda lenta focales, complejos onda aguda onda lenta focales, polipunta focal, polipunta onda lenta focal, duración de onda lenta, amplitud de la punta, amplitud de la onda lenta, duración fase de ascenso de punta u onda aguda, frecuencia del complejo punta onda lenta u onda aguda onda lenta en Hz, presencia de dipolo rolándico típico, punta onda lenta generalizada, polipunta onda lenta generalizada, máxima actividad irritativa en N1, máxima actividad irritativa en N2, máxima actividad irritativa en N3, máxima actividad irritativa en REM, actividad paroxística aislada, actividad paroxística en salvas, número de complejos por salva, índice de actividad en sueño, índice de actividad interictal en vigilia por minuto, presencia de actividad irritativa extra centro temporal, actividad irritativa frontal, actividad irritativa parietal y actividad irritativa occipital.
Variables familiares
Antecedente familiar de epilepsia en padre, madre, hermano o familiar de segundo grado. Antecedente familiar de epilepsia rolándica en padre, madre, hermano o familiar de segundo grado, historia familiar de Trastorno por déficit de atención e hiperactividad (TDAH), historia familiar de trastornos del lenguaje, historia familiar de sonambulismo. Comorbilidades: TDAH, trastorno de lenguaje, bajo rendimiento escolar, repitencia escolar, trastorno oposicionista desafiante (TOD), sonambulismo. Además, si recibía o no tratamiento, si era el caso, con qué medicamento, si estaba controlada la epilepsia o si hubo empeoramiento con el medicamento.
Los datos se registraron en la base de datos del estudio en Excel y finalmente se exportaron los datos para su análisis en el software estadístico SPSS, versión 16.0.
Para aquellas variables de tipo cualitativo se calcularon las frecuencias y proporciones. Para las de tipo cuantitativo se verificó si su distribución era normal, en cuyo caso se presentan como promedios y desviaciones estándar. De lo contrario se presentan las medianas y sus respectivos rangos intercuartiles. En el análisis exploratorio se utilizaron las pruebas de chi cuadrado, test exacto de Fisher y test Shapiro –Wilk.
Aspectos éticos
Este estudio se clasificó en la categoría de investigación sin riesgo, dado que no se realizó evaluación directa de los pacientes. La información obtenida en el estudio sobre cada uno de los participantes se manejó de forma confidencial, con fines académicos y científicos; solo el equipo investigador tuvo acceso a los datos personales de los pacientes. Se solicitó a las correspondientes dependencias de la IPS involucradas, el permiso para la recolección de la información. Se tuvo en cuenta la Declaración de Helsinki y la resolución del Ministerio de Protección Social sobre investigación. Fue aprobado por los Comités de Ética del Instituto de Investigaciones de la universidad de Antioquia y de la IPS Universitaria de Medellín.
RESULTADOS
Se incluyeron en el estudio 44 pacientes, 20 (46%) fueron mujeres y 24 (54%) hombres, el promedio de edad fue 7,7 años (DE 2.3) y la edad de inicio de la epilepsia fue 6,6 años (DE: 2.3). La mayor proporción de pacientes (70%) eran naturales de Medellín y su área metropolitana (Tabla 1).
Respecto a las manifestaciones clínicas de la epilepsia rolándica, se encontró que la proporción de pacientes que presentó crisis solo en sueño (43%) fue similar a la proporción de pacientes que presentó crisis solo en vigilia (46%) y una proporción muy baja presentó crisis tanto en sueño como en vigilia (11%). El 76% de los pacientes había presentado un máximo de 4 crisis en total. Se describen las principales manifestaciones clínicas (Tabla 2).
Solo en 10 pacientes se obtuvo el dato de la lateralidad de la crisis, siendo derecha en el 90% de los casos. En el 64% hubo generalización de la crisis y en el 36% se reportaron crisis con duración mayor a 5 minutos. Solo 1 paciente (2%) avisó el inicio de la crisis.
En la revisión de los electroencefalogramas se encontró que la actividad irritativa se presentaba en proporciones similares en el lado izquierdo y el lado derecho. La actividad irritativa encontrada estuvo representada principalmente por complejos punta-onda lenta (61%) y onda aguda-onda lenta (39%) focales. No se encontró actividad generalizada en ninguno de los pacientes (Tabla 3).
Se realizó la medición de los complejos punta-onda lenta y onda aguda-onda lenta (Tabla 4). En el 93% de los pacientes se encontró dipolo rolándico típico.
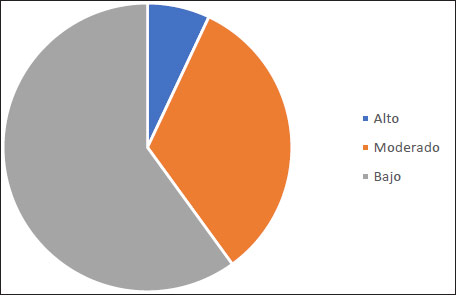
Al evaluar la actividad irritativa en sueño se encontró que esta era máxima en sueño NREM y no se encontró en REM. Las fases del NREM en las que se presentó la actividad irritativa fueron N1 (23%) y N2 (84%). El índice de actividad interictal en sueño fue bajo en el 59% de los pacientes (Figura 1).
La actividad irritativa se presentó aislada en el 43% de los casos y en salvas en el 57%. Cuando se presentó en salvas, éstas estaban compuestas por máximo 5 complejos de punta-onda lenta u onda aguda-onda lenta en el 75% de los casos y solo en el 8% de los casos presentaban más de 10 complejos por salva.
Adicionalmente, se encontró que el 40% no presentaba actividad irritativa en vigilia, y el 60% que si la presentaba podía tener un índice de actividad en vigilia desde 1 hasta 29 por minuto, aunque en el 84% de los pacientes el índice era de 1-2/min.
| Tabla 1. Lugar de origen de los pacientes con epilepsia rolándica (N=44) |

*Apartadó, Carmen de Viboral, Rionegro, San Antonio de Prado, San Pedro de los Milagros, Segovia. |
| Tabla 2. Características clínicas pacientes con epilepsia rolándica (N=44) |
 |
| Figura 1. Índice de actividad interictal en sueño |
 |
Se presentó actividad extra centro-temporal en 9 pacientes (20%), siendo parietal en 5, occipital en 3 y frontal en 1.
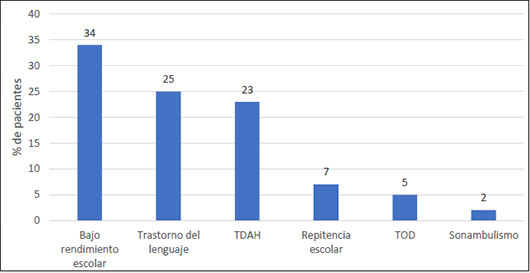
El 32% de los pacientes tenía antecedente familiar de epilepsia, siendo en el 79% de los casos un familiar de segundo grado quien presentaba este antecedente. Solo en el 2% se encontró antecedente familiar de epilepsia rolándica. Otros antecedentes familiares documentados fueron TDAH (5%), trastorno del lenguaje (14%) y sonambulismo (16%). En los pacientes con epilepsia rolándica se encontraron como comorbilidades más frecuentes el bajo rendimiento escolar en 34%, trastorno del lenguaje en 25% y TDAH en 23% (Figura 2).
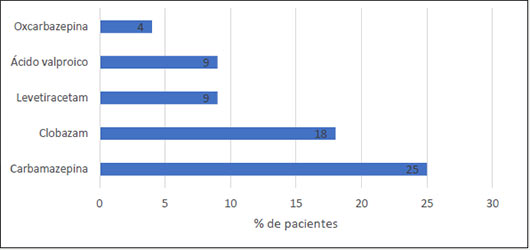
El 61% de los pacientes estaba recibiendo tratamiento farmacológico para la epilepsia, siendo la carbamazepina y el clobazam los medicamentos más usados, seguidos por levetiracetam, ácido valproico y oxcarbazepina (Figura 3). No se reportaron otros medicamentos. Todos los pacientes tenían control de la epilepsia y en ningún caso se reportó empeoramiento de la epilepsia con el tratamiento.
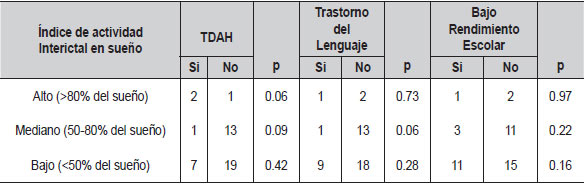
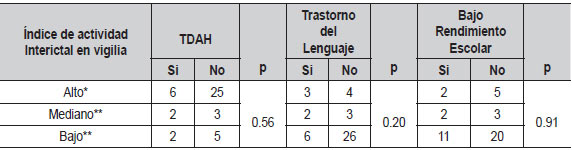
Se realizó un análisis exploratorio intentando determinar si algunas variables clínicas, eléctricas y/o familiares podían influir en la presentación de la epilepsia rolándica. Al evaluar si el índice de actividad interictal en sueño y vigilia se relacionaba con las comorbilidades TDAH, trastorno del lenguaje y bajo rendimiento escolar, no se encontraron diferencias significativas (Tablas 5 y 6).
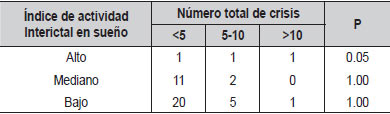
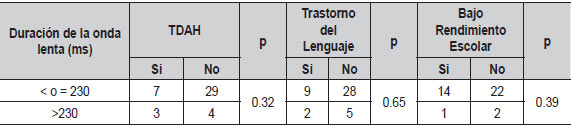
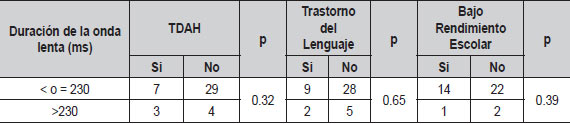
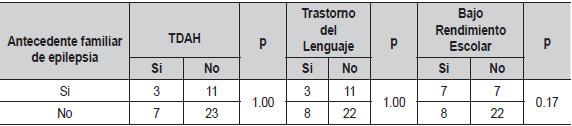
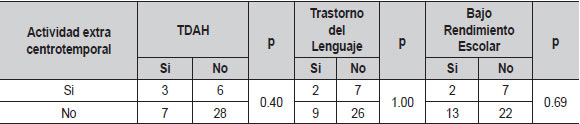
Al evaluar si el índice de actividad interictal en sueño se relacionaba con el número total de crisis, no encontramos diferencias significativas (Tabla 7). Al comparar la duración de la onda lenta, la lateralidad de las puntas centrotemporales, la actividad extra centrotemporal y el antecedente familiar de epilepsia con comorbilidades tampoco hubo diferencias (Tablas 8, 9, 10, 11). El voltaje de la punta u onda aguda no mostró ninguna relación con las principales comorbilidades.
Cuando se revisó la relación entre la frecuencia de los complejos en Hz con las comorbilidades, no hubo diferencias para TDAH y bajo rendimiento escolar. Sin embargo en el caso del trastorno del lenguaje se encontró que la mediana de las frecuencias de los complejos en los pacientes con trastornos del lenguaje era más baja: 3.3 Hz, que en los pacientes sin trastorno del lenguaje: 3.6 Hz (p=0.02) (Figura 4). No hubo diferencias significativas al comparar el antecedente familiar de epilepsia según género (p=1.00).
DISCUSIÓN
Como se ha mencionado previamente, la epilepsia benigna de la infancia con puntas centro- temporales es el síndrome epiléptico más frecuente de la infancia. Se ha descrito una predominancia masculina de 3: 1 (19), pero en nuestro estudio no encontramos diferencias significativas según el género.
Se sabe que este tipo de epilepsia inicia generalmente antes de los 13 años, con un pico de presentación a los 7-9 años (20); sin embargo, si bien en nuestro estudio el inicio de la epilepsia se dio antes de los 13 años, el promedio de edad de presentación fue a los 6,6 años, lo que está ligeramente por debajo de lo reportado.
Característicamente las crisis de la epilepsia rolándica se presentan durante el sueño en más del 50% de los casos y solo en vigilia en el 5-25% (21). En nuestra serie la proporción de crisis en sueño fue similar a la proporción en vigilia.
| Tabla 3. Características electroencefalográficas de pacientes con epilepsia rolándica (N=44) |
|
| Figura 2. Comorbilidades de pacientes con epilepsia rolándica |
 |
En general el número de crisis en este síndrome es bajo, siendo menor a 5 crisis en el 50% (22). En nuestra serie este porcentaje asciende a 76%.
En la revisión de Park et al (2015), se describe la frecuencia de los síntomas: Sonidos faríngeos 53%, disartria 40%, manifestaciones sensoriomotoras faciales 30%, sialorrea 30%. Estas últimas 2 manifestaciones se presentaron con una frecuencia muy similar en nuestro grupo de pacientes, pero la frecuencia de disartria y sonidos faríngeos estuvo casi un 50% por debajo de lo reportado en la literatura. Respecto a la generalización de las crisis que usualmente es cercana al 50%, se presentó en el 64% de nuestro grupo.
| Tabla 4. Medición de los complejos punta-onda lenta y onda aguda -onda lenta |
*Punta-onda lenta u onda aguda-onda lenta |
| Tabla 5. Índice de actividad Interictal en sueño en relación con las principales comorbilidades(N=44) |
 |
| Figura 3. Medicamentos usados para el tratamiento de la epilepsia rolándica |
 |

| Tabla 6. Índice de actividad Interictal en vigilia en relación con las principales comorbilidades(N=44) |
* Alto: >5 complejos/min, **Moderado: 2-5 complejos/min, ***Bajo: < 2complejos/min |
| Tabla 7. Relación del índice de actividad Interictal en sueño con el número total de crisis (N=44) |
 |
| Tabla 8. Relación de duración de la onda lenta con principales comorbilidades (N=44) |
 |
| Tabla 9. Relación de la lateralidad de las puntas centrotemporales con las principales comorbilidades (N=44) |
 |
| Tabla 10. Relación de la actividad extra centrotemporal con principales comorbilidades (N=44) |
|
| Tabla 11. Relación del antecedente familiar de epilepsia con principales comorbilidades (N=44) |
 |
| Figura 4. Frecuencia del complejo (HZ) en pacientes con epilepsia rolándica, con y sin trastorno del lenguaje |
 |
Al comparar las alteraciones electroencefalográficas reportadas en la revisión de Guerrini y Pellacani (2012), con nuestro estudio, podemos ver que en ambos se presenta activación de la actividad en sueño, esta actividad se presenta con frecuencia en salvas, sin formar polipuntas y la actividad irritativa es bilateral en cerca de un tercio de los pacientes.
Además, al evaluar las características electroencefalográficas clásicas de la epilepsia rolándica descritas por Kellaway en el 2000, encontramos que la duración promedio de la punta es de 74 ms, por lo que se considera que en realidad en este síndrome se presentan tanto puntas como ondas agudas, así el nombre del síndrome mencione solo puntas; las cuales son seguidas de onda lenta. En nuestros pacientes las puntas tuvieron duración promedio de 70.4 ms.
La amplitud de la punta es en promedio de 169 uV, llegando incluso hasta 300 uV. En nuestra serie el promedio de la amplitud fue un poco más baja: 122,1 uV.
Al igual que Kellaway, encontramos que la actividad rolándica se presenta principalmente en sueño NREM y es menos frecuente en vigilia. Por otro lado, Blume et al (1984) describió que la frecuencia en Hz de los complejos POL u onda aguda-onda lenta en pacientes con epilepsia rolándica era de 1,5-3 Hz (22) lo que difiere de nuestra serie en la que encontramos frecuencias promedio de 3,87 Hz.
Respecto a los antecedentes familiares de pacientes con epilepsia rolándica se ha reportado que hasta el 10% de los pacientes tienen antecedente familiar de epilepsia (23), lo que contrasta con el 32% en nuestros pacientes. Así también antecedente familiar de trastorno del lenguaje en 17% (24), similar a lo encontrado en nuestro estudio: 14 %. No hay datos respecto al antecedente familiar de TDAH, ni sonambulismo.
Los estudios que han evaluado comorbilidades en niños con epilepsia rolándica han reportado TDAH en 19-68%, bajo rendimiento escolar en el 30% y trastorno del lenguaje en el 29% (24-25). En nuestros pacientes encontramos estas comorbilidades en el 23%, 34% y 25% respectivamente, lo cual es similar a lo reportado en la literatura.
El estudio realizado por Liu et al en 2017, con 1817 pacientes, evaluó el uso de tratamiento anticonvulsivo en este síndrome epiléptico, en- contrando que el 75% recibía tratamiento, siendo los medicamentos más frecuentemente utilizados: Oxcarbazepina 37%, ácido valproico 25% y levetiracetam 19% (26). Dichos hallazgos difieren de lo encontrado en nuestros pacientes, quienes se encontraban en tratamiento farmacológico principalmente con carbamazepina y clobazam, siendo el uso de ácido valproico, levetiracetam y oxcarbazepina menos frecuente.
Sin embargo, todos los pacientes se encontraban controlados, lo que podría sugerir una eficacia similar de los medicamentos utilizados.
Cuando se han evaluado las características electroencefalográficas y clínicas de la epilepsia rolándica no se han encontrado muchos factores que se correlacionen con la forma de presentación, evolución, ni comorbilidades (27). Se plantea en un estudio (28) que la demora en el inicio del tratamiento puede ser un factor que favorece la evolución inadecuada y otro estudio reciente (29) propone que los ripples (80-250 Hz) superimpuestos a las puntas rolándicas en el EEG de superficie son útiles para predecir evolución en epilepsia rolándica. Estos aspectos no fueron valorados en nuestros pacientes.
Dado que existen pocos factores reportados como indicadores de mal pronóstico en epilepsia rolándica, decidimos realizar un análisis exploratorio para evaluar la relación de algunas variables electroencefalográficas con la presencia de comorbilidades y con la frecuencia de las crisis, sin que encontráramos diferencias estadísticamente significativas, excepto cuando evaluamos la frecuencia de los complejos (Hz) con la presencia de trastorno del lenguaje como comorbilidad. La mediana de la frecuencia de los complejos fue menor en los pacientes con trastorno del lenguaje (3,3 Hz) respecto a los pacientes sin trastorno del lenguaje (3,6 Hz) con p= 0,02. Esto no ha sido descrito previamente por lo que sería interesante evaluar esta relación en series más grandes.
Este estudio puede valorarse como un continuo de alteraciones que parten desde un paciente con crisis aisladas hasta otros con crisis frecuentes y alteraciones cognitivas severas que afectan el lenguaje y el aprendizaje. La caracterización del cuadro clínico de la enfermedad se realizó hace más de tres décadas y se encuentran numerosos artículos en este sentido en la literatura. En el momento actual se están investigando los factores de riesgo que pudieran explicar el pronóstico del niño y se está profundizando en los aspectos genéticos.
Debido al hecho de que se trata de un estudio retrospectivo en pacientes atendidos en dos centros, el tamaño de la muestra resulta inadecuado para sacar conclusiones en los hallazgos descriptivos y en la asociación de las variables, aunque hemos revisado estudios similares en otras partes del mundo, aparecidos en el último lustro. Encontramos sin embargo unos datos interesantes que deben profundizarse: por ejemplo, los complejos punta onda lenta con frecuencias más bajas (Hz) estuvieron más relacionados con trastornos del lenguaje; y la frecuente comorbilidad de la epilepsia rolándica con bajo rendimiento escolar, TDAH y trastorno del lenguaje.
Es posible continuar el análisis con pacientes estudiados en otras instituciones a nivel local y nacional, para poder realizar un estudio analítico en el que se evalúen varios factores de riesgo que generen información que ayude en el proceso de toma de decisiones cuando haya preguntas para tratar de explicar mejor este cuadro. Aunque este es un tema frecuente en neurología pediátrica, esta es la primera descripción al respecto en nuestro medio. Para respaldar lo observado en esta muestra de pacientes, se requieren estudios con tamaños de muestra mayores e idealmente con diseño prospectivo para determinar las implicaciones de estos hallazgos.
REFERENCIAS
1. Lundberg S. Rolandic epilepsy: A Neuroradiological, neuropsychological and oromotor study. Comprehensive Summaries of Uppsala Dissertations from the Faculty of Medicine. Acta Univ Upsal: Uppsala University; 2004.
2. Commision on classification and terminology of the International League against Epilepsy. Proposal for revised classification of epilepsies and epileptic syndromes. Epilepsia. 1989; 30: 389-99.
3. Chahine L, Mikati M. Benign pediatric localization- related epilepsies: Part II: Syndromes in childhood. Epileptic Disorders. 2006; 8(4):243-58.
4. Engel J. Report of the ILAE classification core group.
Epilepsia. 2006; 47(9):1558-68.
5. Panayiotopoulos CP. Benign childhood focal seizures and related epileptic syndromes. In: Panayiotopoulos CP, editor. The Epilepsies: Seizures, Syndromes and Management. 1 ed. Oxford: Bladon Medical Publishing; 2005. p. 223-34.
6. Astradsson A, Olafsson E, Ludvigsson P, Björgvinsson H, Hauser WA. Rolandic epilepsy: An incidence study in Iceland. Epilepsia. 1998; 39(8):8846.
7. Perkins FF, Breier J, McManis MH, Castillo E, Whe- less J, McGregor AL, et al. Benign rolandic epilepsy- perhaps not so benign: use of magnetic source imaging as a predictor of outcome. J Child Neurol.
2008; 23(4): 389-93.
8. Bulgheroni S, Franceschetti S, Vago C, Usilla A, Pantaleoni C, D’Arrigo S, et al. Verbal dichotic listening performance and its relationship with EEG features in benign childhood epilepsy with centrotemporal spikes. Epilep Res. 2008; 21 (79): 31-8.
9. Fonseca L, Tedrus G, Campregher AA, Capatto M. Benign childhood epilepsy with centrotemporal spikes: A longitudinal neuropsychological and electroencephalographic study. Clin Neurophysiol.
2008; 119:S84.
10. Legarda S, Jayakar P. Electroclinical significance of rolandic spikes and dipoles in neurodevelopmentally normal children. Electro-encephalogr Clin Neurophysiol. 1995; 95:257-9.
11. Tsai M, Hung K. Topographic mapping and clinical analysis of benign childhood epilepsy with centro- temporal spikes. Brain & Dev. 1998; 27–32.
12. Tavares S, Almeida RM, Figueroa SM, Temudo T.
Epilepsia rolándica. Análisis de las características clínicas, electrofisiológicas, tratamiento y pronóstico en 87 pacientes. Rev Neurol. 2005; 41:327-30.
13. Dos Santos Riesgo R, Jayakar P, Tellechea Rotta N. Benign rolandic epilepsy: clinical and electroencephalographic correlates. Arq Neuropsiq. 2000;
58(3-B):852-61.
14. Kellaway P. The electroencephalographic features of benign centrotemporal (rolandic) epilepsy of childhood. Epilepsia. 2000; 41(8):1053-6.
15. Drury I. EEG in benign and malignant epileptic syndromes of childhood. Epilepsia. 2002; 43(S3):17-26.
16. Neubauer BA. The genetics of rolandic epilepsy.
Epilepsia. 2000; 41(8):10612.
17. Willmore LJ, Ueda Y. Genetics of epilepsy. J Child
Neurol. 2002; 17:S18-S27.
18. Mellish L, Dunkley C, Ferrie C, Pal D. Antiepileptic drug treatment of rolandic epilepsy and Panayiotopoulos syndrome: clinical practice survey and clinical trial feasibility. Arch Dis Child. 2015; 100:62–67.
19. Saeed M, Azam M, Shabbir N, Qamar ShA. Is Benign Childhood epilepsy with centrotemporal spikes always Benign? Iran J Child Neurol. 2014 ; 8(3): 38-43.
20. Park J, Shahid A, Jammoul A. Common Pediatric
Epilepsy Syndromes. Pediat Ann 2015 ;44(2):e30-5.
21. Guerrini R, Pellacani S. Benign childhood focal epilepsies. Epilepsia. 2012; 53(Suppl. 4): 9–18.
22. Blume WT, Young GB, Lemieux JF. EEG morphology of partial epileptic seizures. Electroencephalogr Clin Neurophysiol. 1984; 57(4):295-302.
23. Ma C, Chan K. Benign childhood epilepsy with centrotemporal spikes: a study of 50 Chinese children. Brain & Develop. 2003; p. 390–395.
24. Vega Y, Smith A, Cockerill H, Tang S, Agirre Z, Goyal S. Risk factors for reading disability in rolandic epilepsy families. Epilepsy Behav. 2015; 53: 174–179.
25. Pérez A, López L, García J, Calleja M, Lastaño C, Losada R, Fournier C. Epilepsias benignas de la infancia: dificultades académicas y alteraciones comportamentales. Rev Neurol. 2012; 54 (1): 17-23.
26. Liu M, Su X, Shi X, Wu G, Zhang Y, Gao L, et al.
Clinical features of benign epilepsy of childhood with centrotemporal spikes in chinese children. Medicine.
2017; 96:4.
27. Fonseca L, Tedrus G, De Camargo E, Berretta M, Campregher A, Costa D. Benign childhood epilepsy with centro-temporal spikes: correlation between clinical, cognitive and EEG aspects. Arq Neuropsiquiatr. 2007; 65(3-A):569575.
28. Fenglai X, Dongmei A, Sihan C, Jiechuan R, Dong Z. Clinical and Electroencephalographic (EEG) Features Associated With Refractoriness in Benign Childhood Epilepsy With Centrotemporal Spikes. Journal of Child Neurology. 2015; 1-7.
29. Van Klink N, Van ‘t Klooster M, Leijten F, Jacobs J, Braun K, Zijlmans M. Ripples on rolandic spikes: A marker of epilepsy severity. Epilepsia. 2016; 1–11.
Recibido: 29 de agosto de 2017.
Aceptado: 11 de septiembre de 2017.
Correspondencia: Yuly Mildred Bayona Ovalles yulybayonao@gmail.com