El rol de VEGF en la Angiogénesis fisiológica y tumoral
Jhan Sebastián Saavedra Torres1, Luisa Fernanda Zúñiga Cerón2, Sofía Isabel Freyre Bernal3, Guillermo Wilson Muñoz Ordoñez4, Carolina Salguero5
Resumen
El proceso de angiogénesis, en el cual se desarrollan nuevos vasos sanguíneos a partir de una red vascular existente, requiere de la activación de los receptores en la superficie de las células endoteliales. En sus procesos fisiológicos, como la reparación de heridas, se observa un aumento en la permeabilidad vascular que induce el depósito de proteínas plasmáticas en la matriz extracelular y favorece la reparación y la cicatrización. No obstante, en algunas patologías como el cáncer, desempeña un papel importante para el crecimiento y la diseminación de las células tumorales. Su activación en el tumor permite, pero no garantiza, la expansión tumoral y, por ende, la ausencia de angiogénesis puede limitar el crecimiento. Por eso, se han desarrollado varios inhibidores con el propósito de interferir específicamente en diferentes etapas de este proceso, por ejemplo, el bevacizumab (Avastin) que se distingue como un anticuerpo monoclonal que reconoce y se une al Factor de Crecimiento Endotelial Vascular (por sus siglas en inglés VEGF).
Palabras clave: Factor de crecimiento endotelial vascular (VEGF), angiogénesis fisiológica, angiogénesis patológica, cáncer, metástasis, metaloproteinasas de la matriz (MMP), proliferación celular.
The role of VEGF in Physiological and tumoral angiogénesis
Abstract
Angiogenesis, the cellular process leading to the development of new blood vessels from an existing vascular network, requires activation of surface receptors of normal endothelial cells by signaling molecules such as the Vascular Endothelial Growth Factor (VEGF). During physiological processes of angiogenesis leading to injury repair, an increase of vascular permeability favors deposits of plasma proteins in the extracellular matrix. However, in pathological processes such as cancer, angiogenesis plays an important role in the growth and proliferation of the tumor cells. The activation of angiogenesis in a tumor induces, but does not guarantee, tumor expansion; hence, the absence of angiogenesis may limit the tumor growth. Angiogenesis inhibitors been developed to interfere with different stages of the process. For example, bevacizumab (Avastin) is recognized as a monoclonal antibody binds specifically to VEGF and inhibits its angiogenic function.
Keywords: Vascular Endothelial Growth Factor (VEGF), physiologic angiogenesis, pathologic angiogenesis, cancer, metastases, matrix metalloproteinases (MMP), cellular proliferation._______________________
1. Estudiante de noveno semestre del programa de Medicina, Facultad de Ciencias de la Salud, Universidad del Cauca. Grupo de
Investigación en Salud (GIS). Popayán, Colombia.
2 Estudiante de noveno semestre del programa de Medicina, Facultad de Ciencias de la Salud, Universidad del Cauca. Grupo de
Investigación en Salud (GIS). Popayán, Colombia.
3 MSc. Departamento de Ciencias Fisiológicas, Facultad de Ciencias de la Salud. Universidad del Cauca, Popayán, Colombia.
4 MD., Esp. Cirugía general; Sub Esp. Cirugía vascular. Profesor titular, Departamento de Cirugía General, Facultad de Ciencias de la Salud, Universidad del Cauca.
5 PhD. Departamento de Biología Celular y Molecular, Universidad de Harvard, Cambridge, Massachusetts 02138, USA. Grupo de
Investigación en Salud (GIS).
_______________________
Introducción
El rol de la angiogénesis en el cáncer ha sido una de las temáticas más estudiadas. Judah Folkmann formuló los primeros desarrollos en 1971. Postuló algunos de los principios que direccionan las vías de estudio y las aplicaciones terapéuticas de este proceso (1) incluso hoy. En su estudio, demostró que el crecimiento del tumor depende de la angiogénesis y que si no es capaz de desarrollarla, no crecerá más allá de 2 a 3 mm. Con base en ello, en los últimos 10 a 15 años, los desarrollos en las tecnologías y los recursos se han implementado para investigar la biología de la angiogénesis y sus implicaciones como factor pronóstico o alternativa terapéutica para el tratamiento del cáncer (1, 2).
La angiogénesis contempla la formación de nuevos vasos a partir de redes vasculares preexistentes. Se considera un proceso fisiológico importante en la etapa embrionaria para la vascularización de los tejidos. Después, en la etapa posembrionaria, este proceso se lleva a cabo durante la reparación de los tejidos dañados, la formación y el crecimiento de huesos, de manera específica en las mujeres durante el ciclo menstrual y en el desarrollo de la placenta en el embarazo.
Para que el proceso se lleve a cabo de manera adecuada, debe existir un balance entre los mediadores químicos que la promueven (factores proangiogénicos) y los que la inhiben (factores antiangiogénicos) dentro del microambiente tisular. Además, en ese proceso no sólo participan dichos factores, sino que se requiere de la presencia de otras moléculas como proteínas de matriz extracelular, receptores de moléculas de adhesión y enzimas proteolíticas. Estas últimas participan en la degradación de la lámina basal y de las proteínas de matriz, facilitando que se lleve a cabo la migración de las células endoteliales; mientras que los receptores de moléculas de adhesión participan en las uniones entre esas células (3- 5).
Además de participar en distintos procesos fisiológicos, la angiogénesis también se presenta en desarrollos patológicos como el cáncer. La progresión tumoral incluye cambios en el equilibrio local entre los reguladores positivos y negativos del crecimiento vascular (4, 6). El cáncer pertenece a un grupo de enfermedades caracterizadas por el crecimiento incontrolado y la propagación de células anormales. Si la propagación no se controla, puede causar la muerte. Algunos factores externos como el tabaco, los organismos infecciosos y una dieta poco saludable, sumada a factores internos como mutaciones heredadas, factores genéticos, la alteración de hormonas y algunas condiciones inmunológicas pueden inducir este tipo de manifestaciones. Después del crecimiento celular incontrolado, se lleva a cabo un proceso de migración e invasión de tejidos, denominado “metástasis”. Dicho proceso tumoral requiere de un aporte sanguíneo que le brinde el oxígeno, los nutrientes y los factores de crecimiento necesarios para que las células tumorales puedan encargarse del crecimiento y la migración celular (7- 10). En ese contexto, las estrategias anti-VEGF para el tratamiento de los cánceres se han diseñado con el fin de inhibir la neovascularización. Estudios recientes sugieren que el VEGF puede proteger las células de la apoptosis y aumentar su resistencia a la quimioterapia convencional y a la radioterapia. Sin embargo, la combinación de terapias (anti-VEGF con quimioterapia o radioterapia) son más eficaces contra muchos tipos de tumores, posiblemente porque además de la inhibición de la angiogénesis y el bloqueo del VEGF hace que las células tumorales sean más susceptibles al tratamiento convencional (11, 12).
Metodología
En este estudio, se llevó a cabo una revisión bibliográfica con un margen de tiempo entre el año 1974 y 2016. Se preseleccionaron en total 243 documentos, que fueron evaluados mediante la declaración CONSORT y STROBE. Ambas declaraciones determinan la calidad de la información en los estudios, gracias a las cuales, se eligieron 80 documentos referenciados para en el presente artículo. Este proceso se llevó acabo con un alto rigor metodológico a partir de la búsqueda bibliográfica en las bases de datos Scielo, NEJM, Elsevier, Pub-med, Redalyc, American Cancer Society, Science Direct, Medwave, Nature Reviews, EBSCO, Naxos y bases de datos que ofrecen la Universidad del Cauca y la Universidad de Harvard.La búsqueda se desarrolló utilizando los términos MeSH y DeSC: angiogénesis, factor de crecimiento del endotelio vascular, neoplasia, metaloproteinasas de la matriz extracelular, matriz extracelular y cicatrización de heridas. Para la gestión y organización de la información, se utilizó el programa de libre acceso Mendeley. Se escogieron estudios de tipo ensayo clínico, estudios de cohorte, estudios de intervención, metanálisis, artículos originales y artículos de revisión documental que tuvieran rigor sistemático y que se enfocaran en los aspectos bioquímicos, biológicos, patológicos y experimentales de las innovaciones en la angiogénesis.
La búsqueda se limitó a artículos publicados en el idioma inglés y español. Adicionalmente, se realizaron revisiones de textos, manuales, mapas conceptuales y archivos que tuvieran información relevante y constatada sobre la fisiología de la angiogénesis en la actualidad. Finalmente, se construyó el marco teórico representado en el presente artículo.
Objetivo
Desarrollar una revisión de tema que permita una mejor comprensión de la participación del VEGF en los mecanismos de la angiogénesis fisiológica y patológica en el cáncer.
Resultados
La evidencia científica registrada en los diversos estudios que se han realizado sobre el VEGF demuestra la creación de terapias antiangiogénicas, las cuales amplían la supervivencia de los pacientes con procesos tumorales avanzados y han impulsado el interés en el desarrollo de nuevos inhibidores con el fin de evitar y revertir la metástasis. Por lo tanto, para un mejor entendimiento resulta necesario conocer los procesos fisiológicos de la angiogénesis.
La angiogenia
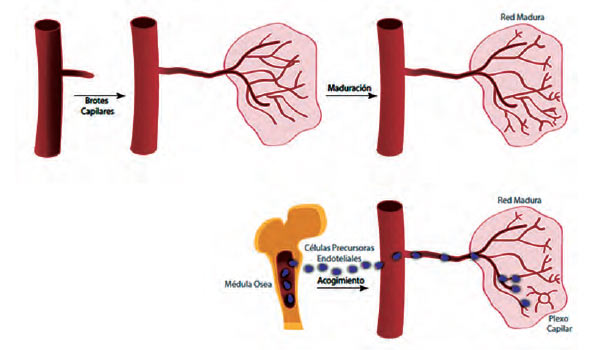
La angiogenia o neovascularización es un proceso fisiológico que ocurre en los humanos durante la adultez. Existen dos mecanismos implicados en este proceso: La angiogenia desarrollada a partir de vasos preexistentes: inicialmente tiene lugar una vasodilatación en respuesta a la presencia de óxido nítrico y el aumento de la permeabilidad gracias al VEGF. Posteriormente, la membrana basal se degrada (proteólisis) por medio de las metaloproteinasas de la matriz (MMP) y la alteración del contacto intercelular entre las células endoteliales por el activador del plasminógeno. Lo anterior, facilita la migración de éstas y su posterior proliferación. Finalmente, ocurre el reclutamiento de las células periendoteliales (pericitos y células musculares lisas vasculares) que dan lugar a la formación de un vaso maduro (13, 14).
La angiogenia a partir de las células precursoras endoteliales (CPE): las CPE se pueden reclutar desde la médula ósea hacia los tejidos para iniciar el proceso. Estas células expresan algunos marcadores de las células madre hematopoyéticas, además de VEGFR-2 y cadherina endotelial vascular (VE-cadherina). Las CPE pueden contribuir a la reendotelialización de los implantes vasculares y la neovascularización de los órganos isquémicos, las heridas cutáneas y los tumores (13,14) (Véase la Figura 1). La angiogenia o neovascularización también se efectúa como un proceso fisiológico.
Moléculas implicadas en la regulación positiva de la angiogénesis
La generación de nuevos capilares es el resultado del balance entre las señales positivas (factores proangiogénicos o estimuladoras) y las señales negativas (factores antiangiogénicos o inhibidores) (15). Existen distintas moléculas implicadas en la regulación positiva de la angiogénesis, incluyendo el factor ácido de crecimiento de fibroblastos (aFGF), el FGF básico, el VEGF, el factor de crecimiento transformante alfa (TGF-α) y el TGF-β, el factor de crecimiento de hepatocitos (HGF), el factor de necrosis tumoral alfa (TNF-α), la angiogenina, la interleucina 8 (IL-8), las angiopoyetinas (Ang-1 y -2), el factor de crecimiento derivado de plaquetas (PDGF), el VEGF derivado de la glándula endocrina (EGVEGF), la leptina, las prostaglandinas, los lípidos, entre otros. Algunos de ellos como el TGF-α y bFGF actúan, al menos en parte, regulando la expresión del VEGF (16,17) (Véase la Tabla 1).
El proceso de pasos múltiples de la angiogénesis incluye la migración y proliferación de células endoteliales (CE), la formación y la organización de grupos celulares en estructuras tubulares, que eventualmente se unirán, para finalmente madurar en forma de vasos sanguíneos estables; aunque debe tenerse presente que existen distintas moléculas implicadas en el proceso (18). En cuanto a los grupos celulares, existen diferentes tipos celulares en el entorno del tumor que contribuyen a la producción de factores que estimulan el proceso de la angiogénesis, entre ellos, las propias células tumorales, las células del estroma y las células del sistema inmune (15).
|
| Figura 1. Angiogenia. Fuente: Adaptado y modificado a partir de los desarrollos de Edward M. Conway. Molecular mechanisms of blood vessel growth. Cardiovasc Res. 2001; 49(1): 507- 21. |
El Factor de Crecimiento Endotelial Vascular (VEGF)
El VEGF, también conocido como factor de permeabilidad vascular (VPF), es producido por muchos tipos de células incluyendo las células tumorales, los macrófagos, las plaquetas, los queratinocitos y las células mesangiales. Desempeña un papel fundamental en las funciones fisiológicas normales, como la formación de hueso, la hematopoyesis, la cicatrización de heridas y el desarrollo de nuevos vasos (11, 12). Asimismo, estimula la supervivencia de las células endoteliales, su proliferación y motilidad, iniciando la gemación de nuevos capilares.
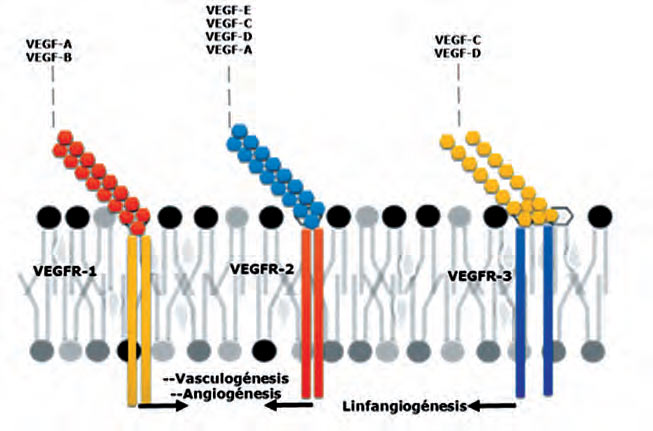
Los miembros de la familia VEGF incluyen proteínas homodiméricas VEGF-A, VEGF-B, VEGF- C, VEGF-D; que realizan su función en las células diana a través de tres receptores con actividad tirosina kinasa intrínseca: VEGFR-1, VEGFR-2 y VEGFR-3, localizados en células endoteliales y en otros tipos celulares (19). Adicionalmente, el VEGF-A regula la angiogénesis y la permeabilidad vascular mediante la activación de dos receptores el VEGFR- 1 y VEGFR- 2. Ambos poseen siete dominios de inmunoglobulinas como dominios extracelulares, una región transmembrana y una secuencia tirosina quinasa que se interrumpe por una quinasa insertada en el dominio (6, 20).
En la misma línea, el VEGFR-1 se expresa preferentemente dentro de la placenta, los trofoblastos situados entre el sistema de los vasos sanguíneos fetales y los maternos.
|
| Tabla 1. Moléculas implicadas en la regulación positiva de la angiogénesis. |

Por lo tanto, se podría considerar como una barrera bioquímica entre la circulación fetal y materna en la placenta mediante la supresión del exceso de la angiogénesis y la permeabilidad vascular anormal (6). Mientras que, el VEGFR-2, más frecuente, participa de manera crucial en la angiogénesis durante el desarrollo y la hematopoyesis, actuando como el mayor mediador de los efectos mitogénicos, angiogénicos y de aumento de la permeabilidad del VEGF (21). El VEGFR- 2 se somete a la dimerización y fosforilación de tirosina dependiente del ligando en células intactas y los resultados en una señal mitogénica, quimiotaxis y pro-supervivencia (20). Finalmente, el VEGFR- 3 pertenece a la misma familia de las RTK (Receptor tirosina quinasa) y al receptor del VEGF-C y el VEGF-D. Se expresa en células endoteliales linfáticas y estimula el receptor de la linfangiogénesis (6, 20). (Véase la Figura 2).
Efectores relacionados con el VEGF y la angiogénesis
Esta proteína desempeña un papel importante en la angiogénesis; entre los muchos factores implicados, el VEGF se ha identificado como uno de los más potentes y predominantes (18) (22). Como su nombre indica, estimula el crecimiento vascular endotelial celular, la supervivencia y la proliferación. En los modelos preclínicos, el VEGF ha demostrado facilitar la supervivencia de los vasos existentes, debido a que contribuye al desarrollo de anomalías vasculares (por ejemplo, tortuosidad e hiperpermeabilidad) que pueden impedir la entrega eficaz de compuestos antitumorales. Se cree que los efectos angiogénicos de la familia VEGF son mediados principalmente a través de la interacción del VEGF con el VE- GFR-2 (23).
|
| Figura 2. Esquema de compatibilidad de unión entre factores de crecimiento y sus receptores. |
También, estimula la permeabilidad vascular en los vasos sanguíneos pequeños (18), promoviendo la fuga vascular con mecanismos de fosforilación de la tirosina de la E-cadherina y β-catenina (24, 25). Las uniones intercelulares, permiten la interacción directa con la célula endotelial vascular, por medio de la unión de complejos de proteínas. Ese proceso permite crear una barrera que es regulada en parte por la estabilidad de adherencias celulares tales como la E-cadherina, β-catenina, p120-catenina y la α-catenina; sustancias que se encuentran implicadas críticamente en el control de la permeabilidad vascular (26).
En la misma línea, el VEGF también promueve la pérdida de esta barrera por la compleja interacción entre los receptores, para los factores de crecimiento angiogénicos que integran las integrinas y las cadherinas permitiendo la fuga de proteínas plasmáticas y la formación de un gel de fibrina extravascular. Esto proporciona un ambiente adecuado para el crecimiento de las células endoteliales. De ese modo, en los tumores, la presencia de altos niveles de VEGF genera que los vasos sean excesivamente permeables y porosos, hecho que aumenta la presión intersticial dentro del tumor y la entrega irregular de nutrientes y oxígeno a este (25, 27). Por otra parte, el VEGF también representa un factor de supervivencia para las células endoteliales, tanto in vitro como in vivo (23, 28). In vitro, el VEGF previene la apoptosis endotelial inducida por la privación de suero. Además, el VEGF produce la expresión de las proteínas antiapoptóticas Bcl-2 (el protooncogén Bcl-2 (B-cell lymphoma 2), A1, XIAP y survivina en las células endoteliales. In vivo, los efectos pro-supervivencia de VEGF se encuentran regulados por el desarrollo (23).Adicionalmente, la tensión de oxígeno desempeña un papel importante en la regulación de la expresión de una variedad de genes (29). La expresión de VEGF es inducida por la exposición baja de presión de O2 en una variedad de circunstancias fisiopatológicas (23). Así pues, la hipoxia constituye el principal factor desencadenante de la angiogénesis. Ésta induce un incremento del factor inducible por la hipoxia (HIF-1) en la célula, que en el núcleo promueve la transcripción de diversos genes, entre los que se encuentran dos potentes factores angiogénicos como: el (VEGF) y el factor de crecimiento endotelial derivado de las plaquetas (PD-ECGF). La unión de estos factores a los receptores de las células endoteliales estimula la fosforilación de distintas proteínas, activándose así las señales de supervivencia para estas células (30).
No obstante, aunque la hipoxia regula la expresión de muchos genes, los mecanismos implicados son poco conocidos. El estudio sobre la actividad del HIF-1 ha demostrado que aumenta la expresión de eritropoyetina (EPO) en respuesta a la hipoxia y, más recientemente, los sitios de unión de HIF-1 se han identificado en los genes que codifican una serie de enzimas glucolíticas. Sin embargo, los mecanismos moleculares por los que la hipoxia regula la expresión de VEGF aún se desconocen (31- 33).
En ese contexto, se sabe que el VEGF se halla presente en todo el ciclo de vida del tumor. A medida que se desarrolla, puede empezar a activar las vías angiogénicas secundarias por medio de la fosforilación de receptores tipo tirosina quinasa. La producción de VEGF se estimula mediante la aparición de flujos de señales externas e internas (en la membrana celular o nuclear), señales ambientales, factores de crecimiento, oncogenes, citoquinas y hormonas. Este proceso es incitado por moléculas como el factor de crecimiento de fibroblastos básico (bFGF), (TGFβ), el factor de crecimiento placentario (PlGF), y el factor de crecimiento de las células endoteliales derivado de plaquetas (PD- ECGF). Como estas vías secundarias emergen, el VEGF continúa siendo expresado, por lo que se reconoce como uno de los mediadores críticos de la angio- génesis (22, 34, 35).
Finalmente, el factor de necrosis tumoral alfa (TNF-a) constituye una citocina inflamatoria con un amplio espectro de actividad biológica, incluyendo la angiogénesis e influye en la formación de nuevos vasos indirectamente. Por otra parte, se ha demostrado que el TNF-α también aumenta la transcripción del gen VEGFR-2 en células endoteliales (36, 37).
Los procesos de señalización intracelular por estimulación con VEGF
Las propiedades citoprotectoras del VEGF exógeno sobre las células endoteliales (CE) descritas hasta ahora incluyen la expresión de las proteínas antiapoptóticas Bcl-2 y survivina, la prostaciclina, la producción de óxido nítrico (NO) y el papel de la vía PIK3K/Akt. La proteína antiapoptótica (B-cell lymphoma 2) Bcl-2 regula la apertura del poro mitocondrial de permeabilidad transitoria inhibiendo la salida del citocromo C al citosol. La proteína Akt inhibe la apoptosis a través de su interacción con las caspasas, el Akt ejerce la regulación del NFKB (nuclear factor-kappa B) y la actividad inhibitoria del p38MAPK (modulador del programa proapoptótico en células endoteliales) permite que las células tumorales expresen inmortalidad para lograr cumplir su objetivo de desarrollar la metástasis, al hacer procesos de expansión por medio de la angiogénesis. A su vez, el Akt es el responsable, en parte, de la producción de NO por VEGF, a través de la fosforilación en Ser/Tre de la NO sintasa endotelial (eNOS) (38, 39).
Adicionalmente, la Fosfoinositol 3-quinasa (PI3k) genera una fosforilación del Akt. El Akt tiene una distribución citoplásmica, pero su fosforilación provoca un reclutamiento hacia la membrana celular. De esta forma, queda expuesto un dominio quinasa que se encontraba oculto en su forma inactiva. Este dominio quinasa de Akt en la membrana se activa mediante dos proteinquinasas de membranas que son la PDK 1 y 2 (40). De este modo, el Akt vuelve al citoplasma en su forma activa y ejerce la fosforilación de distintos efectores relacionados con la supervivencia celular, el aumento del metabolismo de la glucosa y la síntesis de las proteínas (38, 40).
La activación del Akt también puede producirse por una segunda vía, donde participan las integrinas que se conectan a los ligandos de la matriz extracelular. Esto produce la activación de las quinasas, asociadas a las adhesiones focales (FAK) y, estas últimas, actúan sobre el Akt (19, 38). Las adhesiones focales constituyen regiones concretas de la superficie celular en las que se concentran moléculas de adhesión al sustrato (sobre todo integrinas) y numerosas proteínas que conectan estas moléculas al citoesqueleto (19).
La participación de las angiopoyetinas en procesos de angiogénesis
La angiogénesis se encuentra regulada por el VEGF y por las angiopoyetinas que se unen a receptores específicos de las células endoteliales (41). Algunos estudios han determinado la ocurrencia de dos eventos que contribuyen a la adquisición de un fenotipo angiogénico en fases iniciales de la progresión tumoral: la hipoxia y el aumento en la expresión de la angiopoyetina-2 y -1. La hipoxia representa uno de los reguladores fundamentales de los factores que controlan la angiogénesis (42- 44). En esa línea, la angiopoyetina-1 constituye un factor paracrino producido por células de soporte vascular (pericitos), que optimizan la interacción e integración entre estas células y el endotelio. Adicionalmente, permite la formación de vasos poco permeables. Se une de forma específica a un receptor endotelial, el receptor Tie-2 (Tirosina quinasa y dominios homólogos a EGF), y promueve la supervivencia del endotelio y la estabilidad de los vasos. Otro tipo de angiopoyetina, la angiopoyetina-2, puede activar o inhibir la Tie-2 y su expresión se restringe a zonas de remodelación vascular (43, 44). Así pues, es antiangiogénica en ausencia de VEGF y angiogénica en su presencia (45, 46).
En las fases iniciales del desarrollo de los tumores, cuando estos todavía no han alcanzado un tamaño macroscópico, las células tumorales pueden sustraer vasos de los territorios vecinos, pero son incapaces de inducir angiogénesis. Una gran cantidad de angiopoyetina-2, en ausencia de VEGF, mantiene los vasos quiescentes. Sin que se conozca la causa, en una determinada fase del desarrollo del tumor, se expresan concomitantemente la angiopoyetina-2 y el VEGF, lo que dispara la angiogénesis y el crecimiento del tumor. La angiopoyetina-2 también se expresa en los tejidos que experimentan remodelación vascular como la placenta, el útero, el ovario, el testículo inmaduro y el testículo en regresión (41).
Entre los factores reguladores de la angiogénesis, cabe destacar la hipoxia y los estrógenos. Como se ha dicho anteriormente, la hipoxia se reconoce como un factor importante en la inducción de angiogénesis. La falta de oxígeno en el tejido estimula la síntesis del HIF-1α y, a su vez, activa citoquinas proangiogénicas como VEGF y bFGF. Dichos factores promueven la proliferación endotelial y dirigen un gradiente de crecimiento vascular a favor del tumor (14, 41). Por su parte, los estrógenos aumentan la angiogénesis induciendo la expresión de VEGF y del receptor Tie-2. Este receptor, en presencia de angiopoyetina-1, promueve la supervivencia del endotelio vascular. Aunque la unión de la angiopoyetina-2 al receptor Tie-2 provoca apoptosis en ausencia de VEGF, en presencia del factor de crecimiento, aumenta la angiogénesis (41).
Las respuestas de la matriz extracelular en el crecimiento de nuevos vasos sanguíneos
La matriz extracelular (MEC) actúa como un andamio físico para las células y como un repositorio para los factores de crecimiento. La MEC tiene propiedades físico-químicas que transmiten información precisa a las células que influyen profundamente en su biología, por interacciones con receptores de superficie celular denominados integrinas (47). Durante la angiogénesis, la MEC perivascular desempeña un papel crítico en la determinación de las respuestas proliferativas, invasivas y la supervivencia de las células vasculares, guiada por los factores de crecimiento angiogénicos. La interacción dinámica entre el citoesqueleto y la MEC, por contactos focales iniciados por la ligación de integrinas, permiten que las vías de señalización reciban todos los factores de crecimiento necesarios para iniciar cascadas de procesos proliferativos y de supervivencia de las células vasculares (47).
Los complejos de unión permiten que las plaquetas circulantes tengan acceso directo a la lámina subyacente para estimular las células endoteliales (47). La plaqueta se une a los sustratos del proceso que permiten activarla, para que conduzca la descarga de contenidos de sus gránulos alfa que incluyen una variedad de efectores moleculares que varían de proteasa / lipasas a componentes de la MEC y numerosos factores de angiogénesis como por ejemplo: el VEGF y el PDGF (factor de crecimiento derivado de plaquetas) o el S1P (esfingosina-1 fosfato) que se encargan de regular la MEC (48, 49).
La actividad de señalización de la angiogénesis por medio de receptores de tirosina quinasa específicos de angiogénesis depende de la participación de integrinas en la MEC (47). El repertorio de receptores en las células vasculares angiogénicas permite cambios durante la angiogénesis para mantener la interacción celular apropiada con la remodelación de la MEC. Las primeras interacciones ocurren a través de integrinas preexistentes, tales como α vβ5, que se activan corriente abajo por factores de crecimiento y proceden a través de quinasas de la familia Src y Rho quinasa, que facilitan la retracción de las células endoteliales del proceso angiogénico con la liberación de complejos de unión (48, 49).
Los mecanismos de reparación de heridas cutáneas y VEGF
Los mecanismos de reparación de heridas cutáneas se ponen en funcionamiento tras una lesión que altera la continuidad de la superficie. En el proceso, se han identificado tres fases: la inflamatoria, la proliferativa y la de remodelación tisular. Para resaltar al VEGF, se resumirá en el paso de la remodelación tisular que corresponde a la última fase, cuando se desarrolla un tejido estable, similar al existente previo a la lesión, conocido como cicatriz (50).
Las primeras señales para el reclutamiento de los fibroblastos provienen de productos derivados de las plaquetas: el factor de crecimiento derivado de las plaquetas (PDGF), el factor de crecimiento insulinoide (IGF-1) y el factor de crecimiento transformante beta TGF-β. Los monocitos aparacen debido a la atracción que ejercen la fibronectina, la elastina, las quimiocinas, los factores de crecimiento, el IGF-1, el factor de crecimiento nervioso (NGF), TGF-β, la proteína quimio atrayente de macrófagos (MIP)-1α y el VEGF. Los monocitos, con la anterior estimulación en los tejidos, se transforman en macrófagos y actúan removiendo detritus, partículas extrañas y bacterias al igual que los neutrófilos (50, 51).
El mantenimiento de los fibroblastos dentro de la herida se logra a través de señales paracrinas y autocrinas (50). Los macrófagos y los fibroblastos liberan numerosos factores de crecimiento y citocinas que contribuyen a la migración fibroblástica, como por ejemplo: el factor de crecimiento fibroblástico (FGF), IGF-1, (VEGF), IL-1, IL-2, IL-8, PDGF, TGF- a, TGF-b y TNF-a (51). Su presencia predomina entre las 48 a 72 horas después y permanecen por semanas. Asimismo, participan de la última parte de la fase inflamatoria, regulando la llegada de otros monocitos y fibroblastos, liberando factores angiogénicos (proceso favorecido por la hipoxia local) y de crecimiento ((PDGF, FGF, VEGF), TGF-α y β), los cuales son importantes para la migración y proliferación celular y la formación de la matriz extracelular (50). Los fibroblastos que emigran desde el tejido circundante hasta los bordes de la herida son activados por el PDGF y el factor de crecimiento endotelial (EGF), proliferan y empiezan a sintetizar colágeno (51, 52).
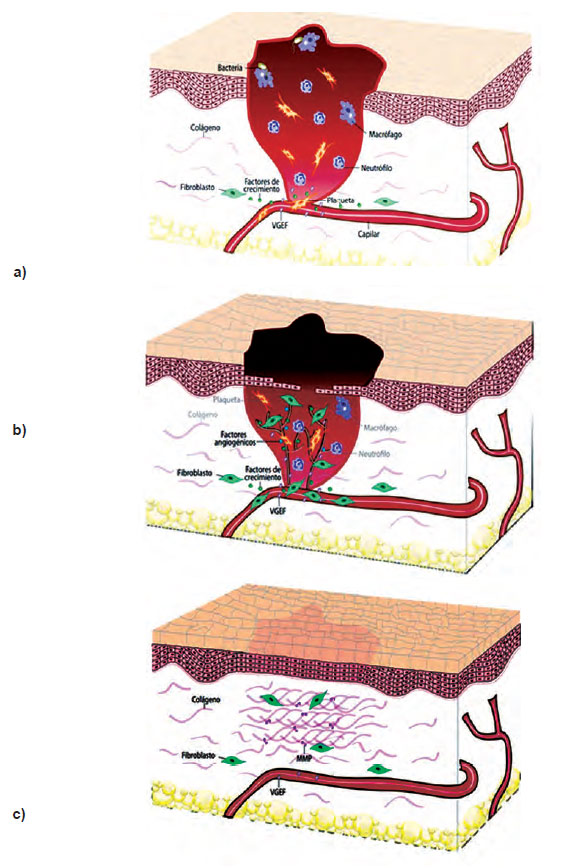
El factor de crecimiento endotelial vascular (VEGF) permite generar la permeabilidad vascular para que los fibroblastos tengan las sustancias necesarias para sintetizar los componentes de la MEC (por ejemplo los colágenos). Luego, la concentración de VEGF disminuye cuando no se necesita más vasos y la MEC se encuentra constituyendo un tejido de cicatrización estable (52, 53). En la Figura 3, se observan: a) Los mecanismos de reparación de heridas cutáneas se ponen en funcionamiento tras una lesión que altera la continuidad de la superficie y se manifiesta en la fase inflamatoria. Para la curación de una herida, se requiere de nuevos vasos de forma que se logre la migración de células y nutrientes que efectúen la reparación. b) La fase proliferativa y la de remodelación tisular, involucran el proceso según el cual el VEGF promueve la angiogénesis en los procesos de inflamación crónica durante la cicatrización, siendo secretado por muchas células del mesénquima y del estroma. c) La fase de remodelación tisular, involucra la conformación de tejido de cicatrización estable.
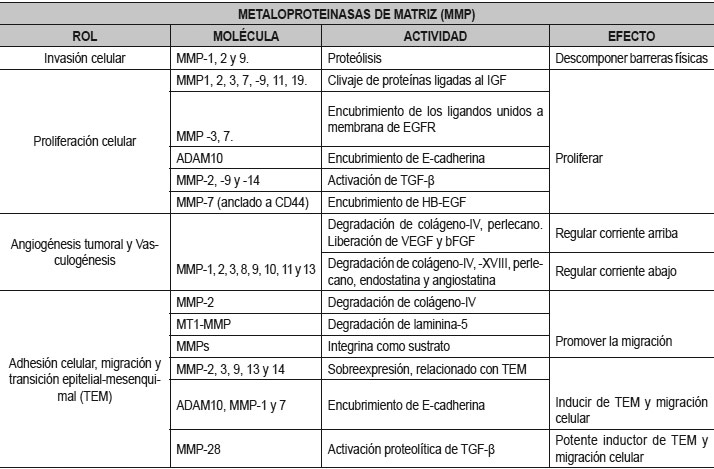
Las MMPs en el proceso de angiogénesis del crecimiento tumoral
Las células del tumor inducen angiogénesis en un proceso similar al de angiogénesis normal. El VEGF, Ang2, y FGF se encuentran implicados en la angiogénesis del tumor, pero en concentraciones más altas que en la angiogénesis de tejido normal. También actúan otros factores que normalmente no están involucrados, como por ejemplo la interleuquina-8 (IL-8), que es una citoquina proinflamatoria. La sobrexpresión de IL-8 en el tumor estimula la producción de metaloproteinasas de matriz 2 (MMP- 2), que degradan la membrana basal, remodelan la matriz extracelular y facilitan la angiogénesis del tumor. La presencia en el torrente sanguíneo de precursores de la célula endotelial CD34+ y de monocitos en áreas de angiogénesis en heridas y tumores, sugiere la hipótesis de que estos precursores hematopoyéticos circulantes podrían contribuir en parte a la regulación de la angiogénesis (1).
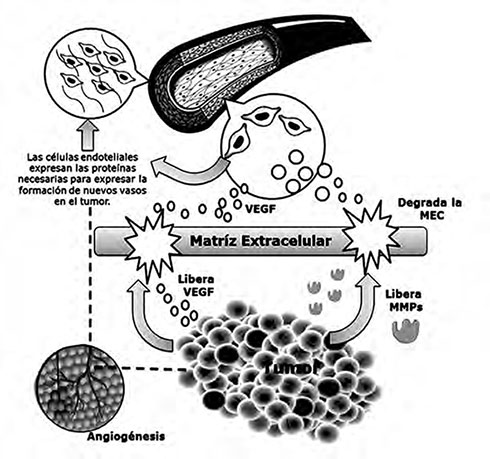
En la Figura 4, se muestra cómo la matriz extracelular es degrada por metaloproteinasas que, liberadas y activadas por el tumor, tienen como objetivo propiciar el crecimiento tumoral, haciendo que las células liberen VEGF y otros factores de crecimiento angiogénico para que migren a las células endoteliales de los vasos más cercanos y que se expresen proteínas necesarias para la formación de nuevos vasos. Eso facilita que luego se regenere la matriz extracelular para nutrir y sostener a los vasos nuevos que permitirán hacer metástasis y aumentar la malignidad del tumor.
Las MMPs representan las principales mediadoras en las alteraciones observadas en el microambiente tumoral durante la progresión del cáncer (54).
|
| Figura 3. Fases de la cicatrización. |
|
| Figura 4. VEGF - en la angiogénesis tumoral. |
Durante el crecimiento tumoral, desempeñan un rol fundamental que consiste en la degradación del tejido conectivo y los componentes de la membrana basal, además de activar los factores de crecimiento, los receptores de superficie para moléculas de adhesión y las quimiocinas (55). Esta interacción con los componentes de la MEC altera la respuesta celular al microambiente, eso permite que las células tumorales se tornen menos adherentes y, por lo tanto, expresan mayor probabilidad de migrar y producir metástasis (56). Durante la carcinogénesis, las células tumorales interaccionan con factores de crecimiento, citoquinas y distintas células, como las células endoteliales, los fibroblastos, macrófagos, mastocitos y pericitos presentes en el microambiente tumoral (57). La capacidad que tienen las células tumorales de migrar, invadir, metastatizar y formar sus propios vasos sanguíneos depende de estas interacciones (58).
La Figura 5 muestra los componentes de una típica cascada de señalización del VEGF. Los mecanismos de la angiogénesis en el tumor, requieren de vasos adicionales para nutrir y permitir la metástasis, con un aumento en la liberación de proteínas y factores de crecimiento endotelial. Los factores de crecimiento pueden ser hormonas o proteínas específicas (VEGF y entre otros factores de crecimiento), que son recibidos por células específicas que interpretan las señales, las cuales a su vez simulan la proliferación celular.
|
| Figura 5. Componentes de una típica cascada de señalización del VEGF. |
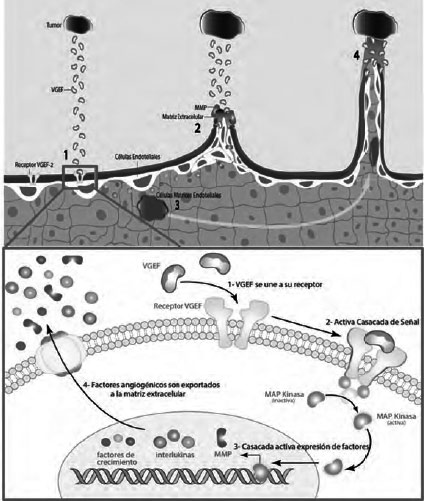
Las vías de señalización patológica de la angiogénesis requieren de un receptor para VEGF y la producción de VEGF aumentada (el tumor libera diversos factores de crecimiento vascular incluyendo VEGF). Cuando la proteína de VEGF se une al receptor (VEGF - 1), activan la vía por medio del uso de la MAP Kinasa que permite la expresión a nivel nuclear de MMP, interleuquinas y factores de crecimiento (el factor de transcripción está conformado por proteínas que se unen al ADN y facilitan la expresión de las moléculas implicadas en la proliferación celular), para luego ser exportados a la matriz extracelular y desencadenar un procesos lítico con MMP. Eso permite abrir el paso a los nuevos vasos y la migración de células precursoras endoteliales hasta el tumor, creando un aumento de intercambio de señales y sustancias para la conexión de los nuevos vasos.
El VEGF controla la señalización y creación de las proteínas necesarias que deben liberar las células precursoras endoteliales al tumor para crear los nuevos vasos, aunque cabe recordar que el VEGF permite inhibir la acción de apoptosis en las células para no interrumpir la angiogénesis que requiere de tiempo y energía a nivel celular. Los nuevos vasos, cuando ya se han comunicado se integran al tumor y posibilitan la expansión tumoral (mitosis descontrolada).
Las MMPs pueden asumir distintos papeles durante la progresión del cáncer dependiendo del estadio del tumor. La Tabla 2 refleja los estadios tempranos, la proteólisis mediada por MMP-3 y -7 de las proteínas específicas que involucran los factores de crecimiento contribuye a la proliferación celular. Posteriormente, el clivaje de las moléculas de adhesión E-cadherina y CD44 activa la motilidad de las células tumorales y facilita las metástasis (58). El MMP-8, en cambio, ejerce un efecto protector disminuyendo el potencial metastásico de las células del cáncer de mama (59). La sobrexpresión de MMP-2 y -9 indica un pronóstico desfavorable al degradar el colágeno tipo IV localizado en las membranas basales e inducir la expresión de factores angiogénicos (60,61).Las MMPs cumplen un rol complejo en la angiogénesis, promoviendo la migración de células endoteliales, liberando VEGF y otros factores proangiogénicos de la MEC como FGF-2 y TGFβ, que también estimulan la proliferación y migración de células endoteliales (62). En modelos animales, se ha demostrado que las MMPs regulan la formación y maduración de nuevos vasos sanguíneos, a través del control que ejercen sobre los factores de crecimiento y citoquinas que actúan en el reclutamiento de pericitos (63). Sin embargo, algunas MMPs pueden tener un efecto inhibidor sobre la angiogénesis, por ejemplo, la hidrólisis del plasminógeno genera fragmentos de angiostatina (64) y la proteólisis del colágeno XVIII da origen a la endostatina (65).Las MMPs no solo degradan la MEC y las membranas basales. Sus acciones son múltiples: modulan la angiogénesis, regulan el curso del proceso inflamatorio y facilitan el reclutamiento de células inflamatorias a través de su acción sobre quimiocinas y citoquinas (66).
|
| Tabla 2. Las MMPs durante la progresión del cáncer. |
El bevacizumab como inhibidor de VEGF
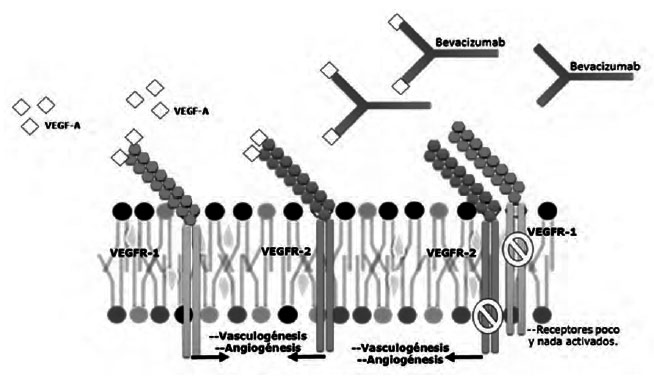
Las células endoteliales son más estables genéticamente que las células del cáncer y tienen un periodo de duplicación de 120 días aproximadamente. La estabilidad genómica junto con su longevidad (comparadas con las de las células tumorales) pueden representar un blanco ideal para las terapias dirigidas contra ellas (18). Las células endoteliales no escaparán a la terapia, pues no experimentarán mitosis con una tasa tan rápida y no desarrollarán ninguna mutación de resistencia a la droga en la generación siguiente (18). En ese contexto, el bevacizumab constituye un anticuerpo monoclonal humanizado que se une al VEGF. Pertenece a una clase de medicamentos conocidos como inhibidores de la angiogénesis (67), actúa evitando la unión a sus receptores Flt-1 (VEGFR-1) y KDR (VEGFR-2), situados en la superficie de las células endoteliales y desacelerando o deteniendo el desarrollo del cáncer. En esa vía, se ha demostrado que encoge o disminuye el crecimiento de los cánceres ováricos epiteliales en etapa avanzada (20, 68- 77).
El bevacizumab fue aceptado en Estados Unidos desde 2004 y, en 2005, por la Agencia Europea de Medicamentos. La buena tolerancia indica que tiene actividad positiva en el tratamiento de cáncer de células renales, cáncer de páncreas y cáncer de mama metastásico (68,69,72) (Véase la Figura 6).
|
| Figura 6. Acción del Bevacizumab. Fuente: Adaptada y modificada de FDA Comunicado de prensa sobre Avastin, recuperada15/04/2014. |
Los estudios clínicos para determinar si el bevacizumab funciona mejor cuando se administra junto con quimioterapia han demostrado buenos resultados en términos de que logra reducir el tamaño de los tumores (o detener el crecimiento de éstos). Sin embargo, no parece ayudar a las mujeres a vivir, aumentando su supervivencia. Cabe señalar que la aplicación del antiangiogénico (anti VEGF) siempre se acompaña con otra terapia de elección como la aplicación de antineoplásicos o antiinflamatorios (6, 8, 68, 69, 72, 78- 80). La sobrexpresión de VEGF es un paso en la generación de metástasis. Sin embargo, la terapias anti-VEGF son importantes para el tratamiento de ciertos cánceres y de la degeneración macular asociada a la edad. Se pueden utilizar anticuerpos monoclonales, tales como bevacizumab (uso exclusivo para cáncer), lapatinib, sunitinib, sorafenib, pazopanib; pequeñas moléculas disponibles para el suministro por vía oral que inhiben las tirosina kinasas estimuladas por VEGF (68, 69, 72).
Conclusiones
- Las células del tumor inducen la angiogénesis mediante un proceso similar al de la angiogénesis normal.
- La producción de VEGF se considera normal siempre y cuando las células no estén activadas y programadas en condiciones oncogénicas, porque cuando lo están la producción de VEGF se expresa en concentraciones mayores, con actividades de modificación citoesquelética y antiapoptosis.
- El VEGF constituye el mediador principal de la angiogénesis en el cáncer. Su expresión es regulada por la expresión de oncogenes, una variedad de factores de crecimiento y la hipoxia
- El VEGF favorece los procesos de angiogénesis fisiológicos como la reparación de heridas y es responsable de un marcado aumento de la permeabilidad vascular, favoreciendo un depósito intenso de proteínas plasmáticas en la matriz extracelular. Además proporciona un estroma provisional para la penetración de los fibroblastos y las células endoteliales, hecho que contribuye en la fase de proliferación.
- Después de una serie de ensayos clínicos en 2004, Avastin fue aprobado por la FDA, convirtiéndose en el primer fármaco antiangiogénico disponible en el mercado.
Agradecimientos
Los autores expresan su agradecimiento al Departamento de Cirugía General de la Universidad del Cauca.
Financiación
Ninguna declarada por los autores.
Conflictos de interés
Ninguno declarado por los autores.
Referencias
1. René. L. Angiogenesis and cancer. Medwave. 2007;
3 (1): 3546.
2. Peter C. Angiogenesis in life, disease and medicine.
Nature. 2005; 438 (1): 932- 6.3.
3. Jiménez-Andrade G y Espinosa C. Inflamación y angiogénesis : el papel facilitador de las células cebadas en el desarrollo del melanoma. Medigraphic.
2011;6 (235): 111- 9.4.
4. Sánchez-Socarrás V. Papel de la angiogénesis en el crecimiento tumoral. Rev Cuba Invest Biomed.
2001; 20 (3): 310.5.
5. Hayden EC. “Cutting off cancer’s supply lines”. Nature. (s.f.); 458 (7239): 686-7. doi:10.1038/458686b. PMID 19360048.
6. Shibuya M. Vascular Endothelial Growth Factor
(VEGF) and Its Receptor (VEGFR) Signaling in Angiogenesis: A Crucial Target for Anti- and Pro- Angiogenic Therapies. Genes Cancer. 2011; 2 (12):
1097- 105.7.
7. Lacoste J, Aprikian AG, Chevalier S. Focal adhesion kinase is required for bombesin-induced prostate cancer cell motility. Mol Cell Endocrinol. 2005 May;
235 (1- 2): 51- 61. Disponible en: https://www.ncbi.nlm.nih.gov/pubmed/15866427
8. American Cancer Society. Cancer Facts and Figures.
1 ed. Atlanta: American Cancer Society: 2015; 3- 7.
9. DeVita VT Jr., Lawrence TS., Rosenberg. SA. Cancer: principles & practice of oncology. 8va ed. Philadelphia: Wolters Kluwer: 2008.10.
10. Gupta GP. Cancer metastasis: building a framework.
Cell. 2006; 127 (1): 679- 95.11.
11. Duffy AM. Vascular Endothelial Growth Factor (VEGF) and Its Role in Non-Endothelial Cells: Autocrine Signalling by VEGF. Firts. Madame Curie Bioscience Database. USA: Bioscience.
12. Reichardt LF. Extracellular matrix molecules and their receptors: Functions in neural development. Annu Rev Neurosci. 1991; 14 (1): 531- 70.13.
13. Edward M. Conway. Molecular mechanisms of blood vessel growth. Cardiovasc Res. 2001; 49 (1): 507-
21.14.
14. Kumar V, Abbas A, Fausto N, Aster JC. Patología Estructural y Funcional Robbins y Cotran. 8va Ed. Barcelona, España: Elsevier Saunders; 2010. 117;
120- 669 pp.
15. Jiménez Cuenca B. Mecanismo de inhibición de la angiogénesis tumoral por trombospondina-1. Nefrología. 2003; 23 (Supl. 3): 49- 53.16.
16. Yancopoulos GD., Davis S., Gale NW., Rudge JS., Wiegand SJ. HJ. Vascular-specific growth factors and blood vessel formation. Nature. 2000; 407 (1): 242- 8.
17. Dvorak HF. Angiogenesis: update 2005. J Thromb
Haemost. 2005; 3 (1):1835- 42.18.
18. Martínez-Hezquerro JD, Herrera LA. Angiogénesis: VEGF/VEGFRs como blancos terapéuticos en el tratamiento contra el cáncer. Cancerología. 2006;
1 (1): 83- 96.19.
19. Thakker GD. HD. The role of phosphatidylinositol
3-kinase in vascular endotelial growth factor signaling. J Biol Chem. 1999; 274 (1): 10002- 7.20.
20. Ferrara N, Gerber PH. The biology of VEGF and its receptors. Nat Med. 2003; 9 (6): 669- 76. Disponible en: http://www.ncbi.nlm.nih.gov/pubmed/12778165
21. Folkman J. DJV. Cáncer: Principios y Práctica de Oncología. Vol 2. 7a Ed. Philadelphia: PA: Lippincott Williams & Wilkins; 2005. 2865- 2882 pp.23.
22. Napoleone Ferrara. Vascular Endothelial Growth Factor: Basic Science and Clinical Progress. Endocr Rev. 2003; 25 (4).
23. Dejana E, Orsenijo F, Lampugnani MG. The role of adherens junctions and VE-cadherin in the control of vascular permeability. J Cell Sci. 2008; 121 (Pt 13):2115- 22.25.
24. Weis SM. CD. Pathophysiological consequences of
VEGF-induced vascular permeability. Nature. 2005;
437 (7058): 497- 504.26.
25. Nelson WJ. Regulation of cell-cell adhesion by the cadherin-catenin complex. Biochem Soc Trans. 2008;
36 (Pt 2): 149- 55.27.
26. Jain RK. Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science. 2005;
307 (5706): 58- 62.28.
27. Alon T. Vascular endothelial growth factor acts as a survival factor for newly formed retinal vessels and has implications for retinopathy of prematurity. Nat Med.
1995; 1 (1): 1024- 8.29.
28. Safran M. KJW. HIF hydroxylation and the mammalian oxygen-sensing pathway. J Clin Invest. 2003; 111 (1):
779- 83.30.
29. Berra E., Ginouves A. PJ. The hipoxia-inductible-factor hydroxylases bring fresh air into hipoxia signalling. EMBO Rep. 2006; 7 (1): 41- 5.31.
30. Liu. Y. Hypoxia Regulates Vascular Endothelial Growth
Factor Gene Expression in Endothelial Cells. Circ Res.
1995; 77 (1): 638- 43.32.
31. Shing Y, Folkman J, Sullivan R, Butterfield C. Heparin affinity: purification of a tumor-derived capillary endothelial cell growth factor. Science. 1984; 223 (1): 1296- 9.33.
32. Folkman J y Klagsbrun M. Angiogenic factors. Science.
1987; 235 (1): 442- 7.
33. Inoue M. VEGF-A has a critical, nonredundant role in angiogenic switching and pancreatic beta cell carcinogenesis. Cancer Cell. 1(2):193- 202.
34. Rini BI. Biology and clinical development of vascular endothelial growth factor-targeted therapy in renal cell carcinoma. J Clin Oncol. 2005; 23 (5): 1028- 43.36.
35. Giraudo E., Primo L., Audero E., Gerber HP. Tumor necrosis factor alfa regula la expresión de receptor vascular-2 del factor de crecimiento endotelial y de su cooperación receptor neuropilina-1 en las células endoteliales vasculares humanas. J Biol Chem. 1998;
273 (1): 22128- 35.37.
36. Hoeben A, Landuyt B. Vascular Endotelial Growth Factor y la angiogénesis. Pharmacol Rev. 2004; 56 (4): 549- 80.38.
37. (agregar autores). Papel del VEGF en la respuesta celular a la agresión. Nefrología. 2003; 23 (Supl 3):
54- 7.39.
38. Gerber HP, McMurtrey AK. Vascular endothelial growth factor regulates endothelial cell survival through the phosphatidylinositol 3’-kinase/Akt signal transduction pathway. Requirement for Flk1/KDR activation. J Biol Chem. 1998; 273 (46): 30336- 43.
39. Brazil DP, Park J. HB. PKB binding proteins. Getting in on the Akt. Cell. 2002; 111: (1): 293- 303.41.
40. Mezquita C. Biología Molecular del Cáncer. Conoc web net. 2001. (Completar referencia)
41. Harris AL. Hypoxia-a key regulatory factor in tumor growth. Nat Rev Cancer. 2002; 2 (1): 38- 47.43.
42. Jones N (completar referencia). Tie receptors: new modulators of angiogenic and lymphangiogenic responses. Nat Rev. 2001; 257 (2).(agregar paginage)
43. Hu B, Cheng SY. Angiopoyetina-2: Desarrollo de inhibidores para la terapia del cáncer. Curr Oncol Rep.
2009; 11 (2): 111- 6.45.
44. Mezquita J, Mezquita B PM and MC. Characterization of a novel form of angiopoietin-2 (Ang-2B) and expression of VEGF and angiopoietin-2 during chicken testicular development and regression. PubMed. 1999;
260: 492- 8.
45. Mezquita J, Mezquita, P, Pau M, Mezquita B, Francone V, Vilagrasa X. Genomic structure and alternative splicing of chicken Angiopoietin-2. PubMed. 2000;
275: 643-51.47.
46. Cheresh D a, Stupack DG. Regulation of angiogenesis: apoptotic cues from the ECM. Oncogene. 2008; 27 (48): 6285- 98. Disponible en: http://www.ncbi.nlm.nih.gov/pubmed/18931694
47. Weis S, Cui J, Barnes L. Endothelial barrier disruption by VEGF-mediated Src activity potentiates tumor cell extravasation and metastasis. J Cell Biol. 2004;167 (1): 223- 9.
48. Eliceiri BP., Klemke R., Stromblad S. CD. Integrin alpha- vbeta3 requirement for sustained mitogen-activated protein kinase activity during angiogenesis. J Cell Biol.
140 (1): 1255- 63.50.
49. Benavides J. Reparación de heridas cutáneas. Rev
Asoc Col Dermatol. 2006; 16 (1): 29- 35.51.
50. Teller P. Fisiología de la cicatrización de la herida: de la lesión a la maduración. Surg Clin N Am, Elsevier España. 89 (1): 599- 610.52.
51. Michael J. FGF and VEGF function in angiogenesis: signalling pathways, biological responses and therapeutic inhibition. TRENDS Pharmacol Sci. 22 (4): 201- 7.
52. Anna-Karin Olsson. AD. VEGF receptor signalling in control of vascular function. Nat Rev Mol Cell Biol. 7 (1): 359- 71.
53. Gialeli C, Theocharis AD. KN. Roles of matrix metalloproteinases in cancer progression and their pharmacological targeting. FEBS. 2011; 278: 16- 27.
54. Van Damme J, Struyf S. Chemokineprotease interaccions in cancer. Semin Cancer Biol. 2004; 14: 201- 8.56.
55. Bourboulia D. Matrix metalloproteinases (MMPs) and tissue inhibitors of metalloproteinases (TIMPs): Positive and negative regulators in tumor cell adhesion. Semin Cancer Biol. 2010; 20: 161- 8.57.
56. Noël A, Jost M. Matrix metalloproteinases at cancer tumor-host interface. Semin Cell Dev Biol. 2008; 19:
52- 60.58.
57. Chabottaux V. Breast cancer progression: insights into multifaceted matrix metalloproteinases. Clin Exp Metastasis. 2007; 24: 647- 56.59.
58. Decock J., Hendrickx W., Vanleeuw U. et al. Plasma MMP1 and MMP8 expression in breast cancer: protective role of MMP8 against lymph node metastasis. BMC Cancer. 2008; 8: 77.60.
59. Egeblad M. WZ. New functions for the matrix metalloproteinases in cancer progression. Nat Rev Cancer.
2002; 2: 161- 74.61.
60. Gonzalez LO, Corte MD, Vásquez J.(completar tres autores más y luego si reportar el et al) Study of matrix metalloproteinases and their tissue inhibitors in ductal in situ carcinomas of the breast. Histopathology. 2008;
53: 403- 15.62.
61. Hua H, Li M, Luo T, Yin Y. Matrix metalloproteinases in tumorigenesis: an evolving paradigm. Cell Mol Life Sci. 2011; 68: 3853- 68.63.
62. Sounni NE, Paye A, Host L. MT-MMPS as regulators of vessel stability associated with angiogenesis. Front Pharmacol. 2011; 2: 111.64.
63. Patterson BC. Angiostatin-converting enzyme activities of human matrilysin (MMP-7) and gelatinase B/ Type IV collagenase (MMP-9). J Biol Chem. 1997; 272: 23- 5.65.
64. Lin HC, Chang JH, Jain S, (completar tres autores más y luego si reportar el et al) et al. Matrilysin cleavage of corneal collagen type XVIII NC1 domain and generation of a 28-kDa fragment. Invest Ophthalmol Vis Sci. 2001;
42: 2517- 24.66.
65. Coronato S, Laguens G. Rol de las metaloproteinasas y sus inhibidores en patología tumoral. Med (Buenos Aires). 2012; 72: 495- 502.67.
66. Villanueva MT. Angiogenesis: Going with the flow. Nat
Rev Cancer. Nature Research. 2016 Nov; 16 (12):
751- 751. Disponible en: http://www.nature.com/doifinder/10.1038/nrc.2016.127
67. American Cancer Society. Anti-angiogenesis
Treatment. American Cancer Society. 2009. p. 3- 10.
68. Montes-Vera MR. Aspectos farmacocinéticos de bevacizumab. Rev Hosp Jua Mex. 2013; 80 (1):
73- 8.70.
69. Carmeliet P, Jain RK. Molecular mechanisms and clinical applications of angiogenesis. Nature. 2011;
473 (7347): 298- 307. Disponible en: http://dx.doi.org/10.1038/nature10144
70. Khosravi Shahi P, del Castillo Rueda A. Angiogénesis neoplásica Neoplastic angiogenesis. An Med Interna.
2008; 25 (7).
71. EMA /An agency of the European Union. AVASTIN® (bevacizumab). European Medicines Agency. Firts. United Kingdom; 2015. p. 1- 5.
72. Meza-Junco J, Montaño-Loza A, Aguayo-González Á.
Bases moleculares del cáncer. 2006; 58 (1): 56- 70.74.
73. Komarova NL, Wodarz D. Cancer as a microevolutionary process. Evol Heal Dis. 2010; (May 2014):1- 17.
74. Acharya Suchitra S. Exploration of the pathogenesis of haemophilic joint arthropathy: Understanding implications for optimal clinical management. Br J Haematol. 2012; 156 (1): 13- 23.76.
75. Camacho Damata X, Cabral P. Angiogénesis Tumoral : estrategias diagnósticas y terapéuticas. Salud Mil. 2012; 31 (1): 1-16.
76. Lee JJ, Lotze MT. Molecular basis of metastasis. N Engl J Med. 2009; 360 (16): 1679.
77. Brooks S a, Lomax-Browne HJ, Carter TM, Kinch CE, Hall DMS. Molecular interactions in cancer cell metastasis. Acta Histochem. Elsevier; 2010 Ene;
112 (1): 3- 25. Disponible en: http://www.ncbi.nlm.nih.gov/pubmed/19162308
78. Qian Q, Zhan P, Yu L, Shi Y, Cheng J, Wei S, et al.
Baseline levels and decrease in serum soluble intercellular adhesion molecule-1 during chemotherapy predict objective response and survival in patients who have advanced non-small-cell lung cancer. Clin Lung Cancer. 2011 Mar; 12 (2): 131- 7.Disponible en: http://www.ncbi.nlm.nih.gov/pubmed/21550560
79. Fontana VJ. Cancer immunology. Cutis. 1974; 13 (5): 717- 8.
Recibido: Marzo 8, 2017
Aceptado: Julio 13, 2017
Correspondencia:
hipocratesjsst@hotmail.com